761077Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharmacokinetics, Pharmacodynamics and Drug
pharmaceutics Review Pharmacokinetics, Pharmacodynamics and Drug–Drug Interactions of New Anti-Migraine Drugs—Lasmiditan, Gepants, and Calcitonin-Gene-Related Peptide (CGRP) Receptor Monoclonal Antibodies Danuta Szkutnik-Fiedler Department of Clinical Pharmacy and Biopharmacy, Pozna´nUniversity of Medical Sciences, Sw.´ Marii Magdaleny 14 St., 61-861 Pozna´n,Poland; [email protected] Received: 28 October 2020; Accepted: 30 November 2020; Published: 3 December 2020 Abstract: In the last few years, there have been significant advances in migraine management and prevention. Lasmiditan, ubrogepant, rimegepant and monoclonal antibodies (erenumab, fremanezumab, galcanezumab, and eptinezumab) are new drugs that were launched on the US pharmaceutical market; some of them also in Europe. This publication reviews the available worldwide references on the safety of these anti-migraine drugs with a focus on the possible drug–drug (DDI) or drug–food interactions. As is known, bioavailability of a drug and, hence, its pharmacological efficacy depend on its pharmacokinetics and pharmacodynamics, which may be altered by drug interactions. This paper discusses the interactions of gepants and lasmiditan with, i.a., serotonergic drugs, CYP3A4 inhibitors, and inducers or breast cancer resistant protein (BCRP) and P-glycoprotein (P-gp) inhibitors. In the case of monoclonal antibodies, the issue of pharmacodynamic interactions related to the modulation of the immune system functions was addressed. It also focuses on the effect of monoclonal antibodies on expression of class Fc gamma receptors (FcγR). Keywords: migraine; lasmiditan; gepants; monoclonal antibodies; drug–drug interactions 1. Introduction Migraine is a chronic neurological disorder characterized by a repetitive, usually unilateral, pulsating headache with attacks typically lasting from 4 to 72 h. -

Current and Prospective Pharmacological Targets in Relation to Antimigraine Action
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Erasmus University Digital Repository Naunyn-Schmiedeberg’s Arch Pharmacol (2008) 378:371–394 DOI 10.1007/s00210-008-0322-7 REVIEW Current and prospective pharmacological targets in relation to antimigraine action Suneet Mehrotra & Saurabh Gupta & Kayi Y. Chan & Carlos M. Villalón & David Centurión & Pramod R. Saxena & Antoinette MaassenVanDenBrink Received: 8 January 2008 /Accepted: 6 June 2008 /Published online: 15 July 2008 # The Author(s) 2008 Abstract Migraine is a recurrent incapacitating neuro- (CGRP1 and CGRP2), adenosine (A1,A2,andA3), glutamate vascular disorder characterized by unilateral and throbbing (NMDA, AMPA, kainate, and metabotropic), dopamine, headaches associated with photophobia, phonophobia, endothelin, and female hormone (estrogen and progesterone) nausea, and vomiting. Current specific drugs used in the receptors. In addition, we have considered some other acute treatment of migraine interact with vascular receptors, targets, including gamma-aminobutyric acid, angiotensin, a fact that has raised concerns about their cardiovascular bradykinin, histamine, and ionotropic receptors, in relation to safety. In the past, α-adrenoceptor agonists (ergotamine, antimigraine therapy. Finally, the cardiovascular safety of dihydroergotamine, isometheptene) were used. The last two current and prospective antimigraine therapies is touched decades have witnessed the advent of 5-HT1B/1D receptor upon. agonists (sumatriptan and second-generation triptans), which have a well-established efficacy in the acute Keywords 5-HT. Antimigraine drugs . CGRP. treatment of migraine. Moreover, current prophylactic Noradrenaline . Migraine . Receptors treatments of migraine include 5-HT2 receptor antagonists, Ca2+ channel blockers, and β-adrenoceptor antagonists. Despite the progress in migraine research and in view of its Introduction complex etiology, this disease still remains underdiagnosed, and available therapies are underused. -

New Drug Update: Not All That Glitters Is Gold Idaho Society of Health‐System Pharmacists 2018 Fall Meeting Sun Valley, Idaho September 30, 2018
9/23/2018 New Drug Update: Not All That Glitters is Gold Idaho Society of Health‐System Pharmacists 2018 Fall Meeting Sun Valley, Idaho September 30, 2018 PRESENTERS: Boise VA Medical Center Idaho State University PGY2 Ambulatory Care Residents: PGY2 Pharmacotherapy Resident: Audra Wilson, PharmD; Kat Liu, PharmD; Kailee Morton, PharmD Nitz Bankova, PharmD St. Luke’s Health System Infectious Disease Pharmacy Fellow: PGY2 Oncology Pharmacy Residents: Benjamin Pontefract, PharmD Andrew Li, PharmD; Amanda Wright, PharmD; Bryce Benson, PharmD PGY2 Mental Health Pharmacy Resident: Samantha Patton, PharmD Disclosures No conflicts of interest to disclose Learning Objectives • Describe clinical scenarios where newly approved medications are beneficial. • Compare newly approved medications with current standards of care. • Identify investigational new drugs and their potential clinical applications. 1 9/23/2018 Overview of Topics • Infectious Disease • Ambulatory Care • Diabetes Mellitus • Women’s Health • Anticoagulation • Pain Management • Oncology • Acute Myeloid Leukemia (AML) Infectious Disease: Plazomicin Delafloxacin Benjamin Pontefract, PharmD Plazomicin (Zemdri®):Overview • Systemic antibiotic in the aminoglycoside family • MoA: disrupts the 30s ribosome to prevent protein synthesis • Concentration dependent bactericidal activity • Approved to treat UTIs caused by E. coli, K. pneumoniae, Proteus spp, and Enterobacter cloaecae 2 9/23/2018 Aminoglycoside Shortcomings Toxicities Resistance • Nephrotoxicity • Decreased cell permeability • Ototoxicity • Altered ribosome binding sites • Therapeutic drug monitoring • AMG‐modifying enzymes (AME) Plazomicin: Efficacy • Study 009: • RCT comparing plazomicin IV to meropenem IV for cUTI caused by enterobactereceae • Plazomicin group was non‐inferior to meropenem group • Study 007: • RCT comparing plazomicin IV to polymyxin E IV for BSI cause by carbapenem‐resistant Enterobacteriaceae (CRE) • Plazomicin was associated with less mortality compared to polymyxin E Connolly L, Achaogen, Inc. -

Anti-Infectives Industry Over the Next 5 Years and Beyond
Bridging the innovation gap... New Drug Futures: Products that could change the pharma market to 2013 and beyond Over 70 pipeline prospects This new major and insightful 450 page in 8 major therapy areas analysis evaluates, compares and contrasts the are analysed in this report prospects for the development compounds that could revolutionise the pharmaceutical Anti-infectives industry over the next 5 years and beyond. Cardiovascular CNS The report provides: Gastrointestinal Detailed background and market context for Metabolic each therapy area covered: Musculoskeletal Addressable patient population Oncology Current treatments Sales drivers Respiratory Sales breakers Future treatments Market dynamics – winners and losers Key drug launches by 2013 Unique sales forecasts by major product to 2013 Over 70 key products assessed Unique evaluation scores for key areas such as novelty of mechanism, clinical data and competition Critical and detailed appraisal of each product‟s research and development Extensive pipeline listings, putting the profiled products into their competitive context The search – and need – for new products has never been greater and what’s in the development pipeline has never generated more interest. That is why this analysis is so important! GLOBAL PHARMA MARKET IN CONTEXT THE Are there too many prophets of doom ready to write-off the research-based pharma industry in the future? Too few novel There is plenty on which to base such anxiety. The research-based industry products and an must achieve a fair price in the face of greater cost control, while the aggressive generic burden of regulation is setting the bar high for successful product sector are taking introduction. -

NCT04179474 Study ID: 3110‐108‐002 Title: A
NCT04179474 Study ID: 3110‐108‐002 Title: A Phase 1b, Two‐Part, Open‐Label, Fixed‐Sequence, Safety, Tolerability and Drug‐Drug Interaction Study Between Single Dose Erenumab or Galcanezumab and Multiple Dose Ubrogepant in Participants with Migraine Protocol Date: 14Aug2019 CONFIDENTIAL Protocol 3110-108-002 Ubrogepant (AGN-241688) Title Page Protocol Title: A Phase 1b, Two-Part, Open-Label, Fixed-Sequence, Safety, Tolerability and Drug-Drug Interaction Study Between Single Dose Erenumab or Galcanezumab and Multiple Dose Ubrogepant in Participants with Migraine Protocol Number: 3110-108-002 Product: Ubrogepant (AGN-241688, MK-1602) Brief Protocol Title: Drug-Drug Interaction Study of Ubrogepant with Erenumab or Galcanezumab Study Phase: 1b Sponsor Name and Legal Registered Address: Allergan Pharmaceuticals International Limited, Clonshaugh Industrial Estate, Coolock, Dublin 17, Ireland US Agent: Allergan Sales, LLC, 2525 Dupont Dr, Irvine, California 92612, USA Regulatory Agency Identifying Number(s): IND #113924 SAE Reporting Fax Number/Email: Business Unit Email Phone Fax Allergan Approval Date: 14 August 2019 1 GRD-CLN-T-01-01 v3.0 CONFIDENTIAL Protocol 3110-108-002 Ubrogepant (AGN-241688) Sponsor Signatories: Date Date The signatures of the sponsor signatories are collected on the protocol approval page. 2 GRD-CLN-T-01-01 v3.0 CONFIDENTIAL Protocol 3110-108-002 Ubrogepant (AGN-241688) Table of Contents Title Page .........................................................................................................................................1 -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -
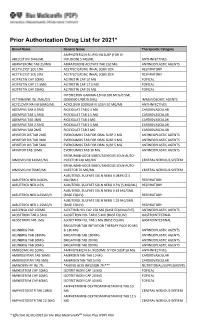
Prior Authorization Drug List for 2021*
Prior Authorization Drug List for 2021* Brand Name Generic Name Therapeutic Category AMPHOTERICIN B LIPID INJ SUSP (FOR IV ABELCET INJ 5MG/ML INFUSION) 5 MG/ML ANTI-INFECTIVES ABIRATERONE TAB 250MG ABIRATERONE ACETATE TAB 250 MG ANTINEOPLASTIC AGENTS ACETYLCYST SOL 10% ACETYLCYSTEINE INHAL SOLN 10% RESPIRATORY ACETYLCYST SOL 20% ACETYLCYSTEINE INHAL SOLN 20% RESPIRATORY ACITRETIN CAP 10MG ACITRETIN CAP 10 MG TOPICAL ACITRETIN CAP 17.5MG ACITRETIN CAP 17.5 MG TOPICAL ACITRETIN CAP 25MG ACITRETIN CAP 25 MG TOPICAL INTERFERON GAMMA-1B INJ 100 MCG/0.5ML ACTIMMUNE INJ 2MU/0.5 (2000000 UNIT/0.5ML) IMMUNOLOGIC AGENTS ACYCLOVIR NA INJ 50MG/ML ACYCLOVIR SODIUM IV SOLN 50 MG/ML ANTI-INFECTIVES ADEMPAS TAB 0.5MG RIOCIGUAT TAB 0.5 MG CARDIOVASCULAR ADEMPAS TAB 1.5MG RIOCIGUAT TAB 1.5 MG CARDIOVASCULAR ADEMPAS TAB 1MG RIOCIGUAT TAB 1 MG CARDIOVASCULAR ADEMPAS TAB 2.5MG RIOCIGUAT TAB 2.5 MG CARDIOVASCULAR ADEMPAS TAB 2MG RIOCIGUAT TAB 2 MG CARDIOVASCULAR AFINITOR DIS TAB 2MG EVEROLIMUS TAB FOR ORAL SUSP 2 MG ANTINEOPLASTIC AGENTS AFINITOR DIS TAB 3MG EVEROLIMUS TAB FOR ORAL SUSP 3 MG ANTINEOPLASTIC AGENTS AFINITOR DIS TAB 5MG EVEROLIMUS TAB FOR ORAL SUSP 5 MG ANTINEOPLASTIC AGENTS AFINITOR TAB 10MG EVEROLIMUS TAB 10 MG ANTINEOPLASTIC AGENTS ERENUMAB-AOOE SUBCUTANEOUS SOLN AUTO- AIMOVIG INJ 140MG/ML INJECTOR 140 MG/ML CENTRAL NERVOUS SYSTEM ERENUMAB-AOOE SUBCUTANEOUS SOLN AUTO- AIMOVIG INJ 70MG/ML INJECTOR 70 MG/ML CENTRAL NERVOUS SYSTEM ALBUTEROL SULFATE SOLN NEBU 0.083% (2.5 ALBUTEROL NEB 0.083% MG/3ML) RESPIRATORY ALBUTEROL NEB 0.5% ALBUTEROL SULFATE -

G Protein-Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. British Journal of Pharmacology (2015) 172, 5744–5869 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: G protein-coupled receptors Stephen PH Alexander1, Anthony P Davenport2, Eamonn Kelly3, Neil Marrion3, John A Peters4, Helen E Benson5, Elena Faccenda5, Adam J Pawson5, Joanna L Sharman5, Christopher Southan5, Jamie A Davies5 and CGTP Collaborators 1School of Biomedical Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK, 2Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK, 3School of Physiology and Pharmacology, University of Bristol, Bristol, BS8 1TD, UK, 4Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK, 5Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2015/16 provides concise overviews of the key properties of over 1750 human drug targets with their pharmacology, plus links to an open access knowledgebase of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. The full contents can be found at http://onlinelibrary.wiley.com/doi/ 10.1111/bph.13348/full. G protein-coupled receptors are one of the eight major pharmacological targets into which the Guide is divided, with the others being: ligand-gated ion channels, voltage-gated ion channels, other ion channels, nuclear hormone receptors, catalytic receptors, enzymes and transporters. These are presented with nomenclature guidance and summary information on the best available pharmacological tools, alongside key references and suggestions for further reading. -

Rimegepant in the Treatment of Migraine
Arch Intern Med Res 2020; 3(2): 119-123 DOI: 10.26502/aimr.0030 Review Article Rimegepant in the Treatment of Migraine Muhammad Adnan Haider1*, Muhammad Hanif2, Mukarram Jamat Ali3, Muhammad Umer Ahmed4, Sundas2, Amin H Karim5 1Department of Internal Medicine, Allama Iqbal Medical College, Lahore, Pakistan 2Department of Internal Medicine, Khyber Medical College Peshawar, Pakhtunkhwa, Pakistan 3Department of Internal Medicine, King Edward Medical University Lahore, Lahore, Pakistan 4Department of Internal Medicine, Ziauddin University and hospital, Karachi, Pakistan 5Associate Clinical Professor of Cardiology, Baylor College of medicine, Houston, United States *Corresponding author: Muhammad Adnan Haider, Department of Internal Medicine, Allama Iqbal Medical College, Lahore, Pakistan, E-mail: [email protected] Received: 02 May 2020; Accepted: 18 May 2020; Published: 21 May 2020 Citation: Muhammad Adnan Haider, Muhammad Hanif, Mukarram Jamat Ali, Muhammad Umer Ahmed, Sundas, Amin H Karim. Rimegepant in the Treatment of Migraine. Archives of Internal Medicine Research 3 (2020): 119- 123. Abstract Migraine is a common and chronic disorder with ent of migraine. This article is focused on exploring the significant financial and socioeconomic burden. It is the role of rimegepant (CGRP-receptor antagonist) by 2nd most common reason for the years lived with reviewing myriads of articles published in Pubmed and disability after back pain. Current available treatment of Google scholar. migraine is limited to subpopulation due to poor tolerability, efficacy, side effects, contraindication and Keywords: Rimegepant; CGRP; Triptans; Migraine; drug-drug interactions. So there is need to evolve some Receptor antagonist treatment to overcome these limitations. Calcitonin- gene-related peptide receptor has been identified in the 1. -

761119Orig1s000 SUMMARY REVIEW
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 761119Orig1s000 SUMMARY REVIEW Summary Review Summary Review Date February 21, 2020 Heather Fitter, MD From Nick Kozauer, MD Billy Dunn, MD Subject Summary Review BLA # 761119 Applicant Lundbeck Seattle BioPharmaceuticals, Inc. Date of Submission December 26, 2018 PDUFA Goal Date February 21, 2020 Proprietary Name Vyepti Established or Proper Names Eptinezumab Solution for intravenous (IV) injection (100 Dosage Form mg/mL) Applicant Proposed Prevention of migraine in adults Indication/Population 100 mg IV infusion every 3 months; 300 mg IV Applicant Proposed Dosing Regimen(s) infusion every 3 months Recommendation on Regulatory Action Approval Recommended Indication/Population Preventive treatment of migraine in adults 100 mg IV infusion every 3 months; 300 mg IV Recommended Dosing Regimen(s) infusion every 3 months 1 Reference ID: 4564908 Summary Review 1. Benefit-Risk Assessment Benefit-Risk Assessment Framework Benefit-Risk Integrated Assessment Eptinezumab is a humanized monoclonal antibody that binds to the calcitonin gene-related peptide (CGRP) ligand, and prevents CGRP binding to its receptor. The applicant provided information supporting safety and efficacy in patients with both chronic migraine (i.e., at least 15 headache days/month, with features of migraine headache on at least 8 days/month) and episodic migraine (i.e., up to 14 migraine headache days/month). There are several FDA-approved drugs for the preventive treatment of migraine. Three drugs in the anti-CGRP class were approved recently for this indication: erenumab (May 2018), fremanezumab (September 2018), and galcanezumab (September 2018). These three drugs are given by subcutaneous (SC) administration either monthly or quarterly, while eptinezumab is given intravenously (IV) quarterly. -

1 Advances in Therapeutic Peptides Targeting G Protein-Coupled
Advances in therapeutic peptides targeting G protein-coupled receptors Anthony P. Davenport1Ϯ Conor C.G. Scully2Ϯ, Chris de Graaf2, Alastair J. H. Brown2 and Janet J. Maguire1 1Experimental Medicine and Immunotherapeutics, Addenbrooke’s Hospital, University of Cambridge, CB2 0QQ, UK 2Sosei Heptares, Granta Park, Cambridge, CB21 6DG, UK. Ϯ Contributed equally Correspondence to Anthony P. Davenport email: [email protected] Abstract Dysregulation of peptide-activated pathways causes a range of diseases, fostering the discovery and clinical development of peptide drugs. Many endogenous peptides activate G protein-coupled receptors (GPCRs) — nearly fifty GPCR peptide drugs have been approved to date, most of them for metabolic disease or oncology, and more than 10 potentially first- in-class peptide therapeutics are in the pipeline. The majority of existing peptide therapeutics are agonists, which reflects the currently dominant strategy of modifying the endogenous peptide sequence of ligands for peptide-binding GPCRs. Increasingly, novel strategies are being employed to develop both agonists and antagonists, and both to introduce chemical novelty and improve drug-like properties. Pharmacodynamic improvements are evolving to bias ligands to activate specific downstream signalling pathways in order to optimise efficacy and reduce side effects. In pharmacokinetics, modifications that increase plasma-half life have been revolutionary. Here, we discuss the current status of peptide drugs targeting GPCRs, with a focus on evolving strategies to improve pharmacokinetic and pharmacodynamic properties. Introduction G protein-coupled receptors (GPCRs) mediate a wide range of signalling processes and are targeted by one third of drugs in clinical use1. Although most GPCR-targeting therapeutics are small molecules2, the endogenous ligands for many GPCRs are peptides (comprising 50 or fewer amino acids), which suggests that this class of molecule could be therapeutically useful. -

Annexes of the Annual Report 2011
1 June 2012 EMA/363033/2012 Office of the Executive Director Annexes of the annual report 2011 The main body of this report is available on the website of the European Medicines Agency (EMA) here. 7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7418 8416 E-mail [email protected] Website www.ema.europa.eu An agency of the European Union © European Medicines Agency, 2012. Reproduction is authorised provided the source is acknowledged. Table of contents Annex 1 – Members of the Management Board ................................................... 3 Annex 2 – Members of the Committee for Medicinal Products for Human Use ......... 5 Annex 3 – Members of the Committee for Medicinal Products for Veterinary Use .... 9 Annex 4 – Members of the Committee for Orphan Medicinal Products.................. 11 Annex 5 – Members of the Committee on Herbal Medicinal Products ................... 13 Annex 6 – Members of the Paediatric Committee .............................................. 16 Annex 7 – Members of the Committee for Advanced Therapies ........................... 18 Annex 8 – National competent authority partners ............................................. 20 Annex 9 – Budget summaries 2010–2011........................................................ 31 Annex 10 – Establishment plan ...................................................................... 32 Annex 11 – CHMP opinions in 2011 on medicinal products for human use ............ 33 Annex 12 – CVMP opinions in 2011