NCT04179474 Study ID: 3110‐108‐002 Title: A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharmacokinetics, Pharmacodynamics and Drug
pharmaceutics Review Pharmacokinetics, Pharmacodynamics and Drug–Drug Interactions of New Anti-Migraine Drugs—Lasmiditan, Gepants, and Calcitonin-Gene-Related Peptide (CGRP) Receptor Monoclonal Antibodies Danuta Szkutnik-Fiedler Department of Clinical Pharmacy and Biopharmacy, Pozna´nUniversity of Medical Sciences, Sw.´ Marii Magdaleny 14 St., 61-861 Pozna´n,Poland; [email protected] Received: 28 October 2020; Accepted: 30 November 2020; Published: 3 December 2020 Abstract: In the last few years, there have been significant advances in migraine management and prevention. Lasmiditan, ubrogepant, rimegepant and monoclonal antibodies (erenumab, fremanezumab, galcanezumab, and eptinezumab) are new drugs that were launched on the US pharmaceutical market; some of them also in Europe. This publication reviews the available worldwide references on the safety of these anti-migraine drugs with a focus on the possible drug–drug (DDI) or drug–food interactions. As is known, bioavailability of a drug and, hence, its pharmacological efficacy depend on its pharmacokinetics and pharmacodynamics, which may be altered by drug interactions. This paper discusses the interactions of gepants and lasmiditan with, i.a., serotonergic drugs, CYP3A4 inhibitors, and inducers or breast cancer resistant protein (BCRP) and P-glycoprotein (P-gp) inhibitors. In the case of monoclonal antibodies, the issue of pharmacodynamic interactions related to the modulation of the immune system functions was addressed. It also focuses on the effect of monoclonal antibodies on expression of class Fc gamma receptors (FcγR). Keywords: migraine; lasmiditan; gepants; monoclonal antibodies; drug–drug interactions 1. Introduction Migraine is a chronic neurological disorder characterized by a repetitive, usually unilateral, pulsating headache with attacks typically lasting from 4 to 72 h. -

Commercial and Medicaid Formulary Changes Effective 1/1/21
Commercial and Medicaid formulary changes effective 1/1/21 These additions and changes apply to Commercial and Medicaid formularies and are effective 1/1/21 unless specified below. Additions Tecartus™ (brexucabtagene autoleucel) – Medical Benefit, PA Required (Reviewed by Medical Director). Rukobia®(fostemavir) – Non-Preferred Brand, QL Required. Phexxi (lactic acid, citric acid, and potassium bitartrate) – Non-Preferred Brand, QL Required. Viltepso (viltolarsen) – Medical Benefit, PA Required. Dayvigo (lemborexant) – Non-Preferred Brand, PA and QL Required. Barhemsys (amisulpride) – Medical Benefit. Blenrep (belanta mabmafodotin-blmf) – Medical Benefit, PA Required. Monjuvi (tafasitamab-cxix) – Medical Benefit, PA Required. Trijardy (empagliflozin/linagliptin/metformin) – Non-Preferred Brand, Step Therapy Required. Procysbiv (cysteamine) – Non-Preferred Brand, PA Required. Hemadyv (dexamethasone) – Non-Preferred Brand, PA and QL Required. Kynmobi (apomorphine) – Non-Preferred Brand, PA and QL Required. Zerviate (cetirizine) – Non-Preferred Brand, Step Therapy Required. Freestyle Libre 2 – Preferred Brand, PA and QL Required. Changes Belsomra (suvorexant) – Change to Non-Preferred Brand, PA and QL Required. Apokyn (apomorphine SC) – Change to Non-Preferred Brand, PA and QL Required. Copaxone (brand only) – Changed from Tier 3 to Tier 2 (only applies to commercial). Humalog (insulin lispro) – Changed from Tier 3 to Tier 2 (only applies to commercial). Nurtec ODT (rimegepant) – Changed from Tier 4 to Tier 3 (only applies to commercial). Reyvow (lasmiditan) – Change to Non-Preferred Brand, PA and QL Required (only applies to commercial). Ubrelvy (ubrogepant) – Change to Non-Preferred Brand, PA and QL Required (only applies to commercial). Soliqua (insulin glargine-lixisenatide) – Changed from Tier 4 to Tier 3 (only applies to commercial). Prefest (estradiol-norgestimate) – Moved to Non-formulary (only applies to commercial). -

New Drug Update: Not All That Glitters Is Gold Idaho Society of Health‐System Pharmacists 2018 Fall Meeting Sun Valley, Idaho September 30, 2018
9/23/2018 New Drug Update: Not All That Glitters is Gold Idaho Society of Health‐System Pharmacists 2018 Fall Meeting Sun Valley, Idaho September 30, 2018 PRESENTERS: Boise VA Medical Center Idaho State University PGY2 Ambulatory Care Residents: PGY2 Pharmacotherapy Resident: Audra Wilson, PharmD; Kat Liu, PharmD; Kailee Morton, PharmD Nitz Bankova, PharmD St. Luke’s Health System Infectious Disease Pharmacy Fellow: PGY2 Oncology Pharmacy Residents: Benjamin Pontefract, PharmD Andrew Li, PharmD; Amanda Wright, PharmD; Bryce Benson, PharmD PGY2 Mental Health Pharmacy Resident: Samantha Patton, PharmD Disclosures No conflicts of interest to disclose Learning Objectives • Describe clinical scenarios where newly approved medications are beneficial. • Compare newly approved medications with current standards of care. • Identify investigational new drugs and their potential clinical applications. 1 9/23/2018 Overview of Topics • Infectious Disease • Ambulatory Care • Diabetes Mellitus • Women’s Health • Anticoagulation • Pain Management • Oncology • Acute Myeloid Leukemia (AML) Infectious Disease: Plazomicin Delafloxacin Benjamin Pontefract, PharmD Plazomicin (Zemdri®):Overview • Systemic antibiotic in the aminoglycoside family • MoA: disrupts the 30s ribosome to prevent protein synthesis • Concentration dependent bactericidal activity • Approved to treat UTIs caused by E. coli, K. pneumoniae, Proteus spp, and Enterobacter cloaecae 2 9/23/2018 Aminoglycoside Shortcomings Toxicities Resistance • Nephrotoxicity • Decreased cell permeability • Ototoxicity • Altered ribosome binding sites • Therapeutic drug monitoring • AMG‐modifying enzymes (AME) Plazomicin: Efficacy • Study 009: • RCT comparing plazomicin IV to meropenem IV for cUTI caused by enterobactereceae • Plazomicin group was non‐inferior to meropenem group • Study 007: • RCT comparing plazomicin IV to polymyxin E IV for BSI cause by carbapenem‐resistant Enterobacteriaceae (CRE) • Plazomicin was associated with less mortality compared to polymyxin E Connolly L, Achaogen, Inc. -
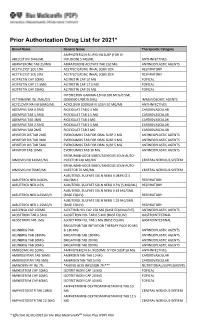
Prior Authorization Drug List for 2021*
Prior Authorization Drug List for 2021* Brand Name Generic Name Therapeutic Category AMPHOTERICIN B LIPID INJ SUSP (FOR IV ABELCET INJ 5MG/ML INFUSION) 5 MG/ML ANTI-INFECTIVES ABIRATERONE TAB 250MG ABIRATERONE ACETATE TAB 250 MG ANTINEOPLASTIC AGENTS ACETYLCYST SOL 10% ACETYLCYSTEINE INHAL SOLN 10% RESPIRATORY ACETYLCYST SOL 20% ACETYLCYSTEINE INHAL SOLN 20% RESPIRATORY ACITRETIN CAP 10MG ACITRETIN CAP 10 MG TOPICAL ACITRETIN CAP 17.5MG ACITRETIN CAP 17.5 MG TOPICAL ACITRETIN CAP 25MG ACITRETIN CAP 25 MG TOPICAL INTERFERON GAMMA-1B INJ 100 MCG/0.5ML ACTIMMUNE INJ 2MU/0.5 (2000000 UNIT/0.5ML) IMMUNOLOGIC AGENTS ACYCLOVIR NA INJ 50MG/ML ACYCLOVIR SODIUM IV SOLN 50 MG/ML ANTI-INFECTIVES ADEMPAS TAB 0.5MG RIOCIGUAT TAB 0.5 MG CARDIOVASCULAR ADEMPAS TAB 1.5MG RIOCIGUAT TAB 1.5 MG CARDIOVASCULAR ADEMPAS TAB 1MG RIOCIGUAT TAB 1 MG CARDIOVASCULAR ADEMPAS TAB 2.5MG RIOCIGUAT TAB 2.5 MG CARDIOVASCULAR ADEMPAS TAB 2MG RIOCIGUAT TAB 2 MG CARDIOVASCULAR AFINITOR DIS TAB 2MG EVEROLIMUS TAB FOR ORAL SUSP 2 MG ANTINEOPLASTIC AGENTS AFINITOR DIS TAB 3MG EVEROLIMUS TAB FOR ORAL SUSP 3 MG ANTINEOPLASTIC AGENTS AFINITOR DIS TAB 5MG EVEROLIMUS TAB FOR ORAL SUSP 5 MG ANTINEOPLASTIC AGENTS AFINITOR TAB 10MG EVEROLIMUS TAB 10 MG ANTINEOPLASTIC AGENTS ERENUMAB-AOOE SUBCUTANEOUS SOLN AUTO- AIMOVIG INJ 140MG/ML INJECTOR 140 MG/ML CENTRAL NERVOUS SYSTEM ERENUMAB-AOOE SUBCUTANEOUS SOLN AUTO- AIMOVIG INJ 70MG/ML INJECTOR 70 MG/ML CENTRAL NERVOUS SYSTEM ALBUTEROL SULFATE SOLN NEBU 0.083% (2.5 ALBUTEROL NEB 0.083% MG/3ML) RESPIRATORY ALBUTEROL NEB 0.5% ALBUTEROL SULFATE -

VYEPTI™ (EPTINEZUMAB) Policy Number: CSLA2020D0090A Effective Date: TBD
Proprietary Information of United Healthcare: The information contained in this document is proprietary and the sole property of United HealthCare Services, Inc. Unauthorized copying, use and distribution of this information are strictly prohibited. Copyright 2020 United HealthCare Services, Inc. UnitedHealthcare® Community Plan Medical Benefit Drug Policy VYEPTI™ (EPTINEZUMAB) Policy Number: CSLA2020D0090A Effective Date: TBD Table of Contents Page Commercial Policy APPLICATION ...................................................... 1 Vyepti™ (Eptinezumab) COVERAGE RATIONALE ........................................ 1 APPLICABLE CODES ............................................. 2 BACKGROUND ................................................... 43 CLINICAL EVIDENCE ............................................ 4 U.S. FOOD AND DRUG ADMINISTRATION ............ 54 CENTERS FOR MEDICARE AND MEDICAID SERVICES 54 REFERENCES ....................................................... 5 POLICY HISTORY/REVISION INFORMATION ...... 65 INSTRUCTIONS FOR USE .................................... 65 APPLICATION This Medical Benefit Drug Policy only applies to state of Louisiana. COVERAGE RATIONALE Vyepti has been added to the Review at Launch program. Please reference the Medical Benefit Drug Policy titled Review at Launch for New to Market Medications for additional details. Chronic Migraine Vyepti is proven and medically necessary for the preventive treatment of chronic migraines when all of the following criteria are met: For initial therapy, all of the -

761119Orig1s000 SUMMARY REVIEW
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 761119Orig1s000 SUMMARY REVIEW Summary Review Summary Review Date February 21, 2020 Heather Fitter, MD From Nick Kozauer, MD Billy Dunn, MD Subject Summary Review BLA # 761119 Applicant Lundbeck Seattle BioPharmaceuticals, Inc. Date of Submission December 26, 2018 PDUFA Goal Date February 21, 2020 Proprietary Name Vyepti Established or Proper Names Eptinezumab Solution for intravenous (IV) injection (100 Dosage Form mg/mL) Applicant Proposed Prevention of migraine in adults Indication/Population 100 mg IV infusion every 3 months; 300 mg IV Applicant Proposed Dosing Regimen(s) infusion every 3 months Recommendation on Regulatory Action Approval Recommended Indication/Population Preventive treatment of migraine in adults 100 mg IV infusion every 3 months; 300 mg IV Recommended Dosing Regimen(s) infusion every 3 months 1 Reference ID: 4564908 Summary Review 1. Benefit-Risk Assessment Benefit-Risk Assessment Framework Benefit-Risk Integrated Assessment Eptinezumab is a humanized monoclonal antibody that binds to the calcitonin gene-related peptide (CGRP) ligand, and prevents CGRP binding to its receptor. The applicant provided information supporting safety and efficacy in patients with both chronic migraine (i.e., at least 15 headache days/month, with features of migraine headache on at least 8 days/month) and episodic migraine (i.e., up to 14 migraine headache days/month). There are several FDA-approved drugs for the preventive treatment of migraine. Three drugs in the anti-CGRP class were approved recently for this indication: erenumab (May 2018), fremanezumab (September 2018), and galcanezumab (September 2018). These three drugs are given by subcutaneous (SC) administration either monthly or quarterly, while eptinezumab is given intravenously (IV) quarterly. -

A Multicenter, Randomized, Open-Label Extension Study to Evaluate the Long-Term Safety and Tolerability of Oral Ubrogepant in Th
NCT02873221 Study ID: UBR‐MD‐04 Title: A Multicenter, Randomized, Open‐Label Extension Study to Evaluate the Long‐Term Safety and Tolerability of Oral Ubrogepant in the Acute Treatment of Migraine With or Without Aura Protocol Amd 3 Date: 11‐Apr‐2018 Allergan Confidential Protocol UBR-MD-04 Amendment 3 Title Page ALLERGAN – CONFIDENTIAL The following contains confidential, proprietary information which is the property of Allergan A MULTICENTER, RANDOMIZED, OPEN-LABEL EXTENSION STUDY TO EVALUATE THE LONG-TERM SAFETY AND TOLERABILITY OF ORAL UBROGEPANT IN THE ACUTE TREATMENT OF MIGRAINE WITH OR WITHOUT AURA Protocol Number: UBR-MD-04 Amendment 3 Phase: 3 Name of Investigational Product: Ubrogepant Sponsor: Allergan Pharmaceuticals Authorized US Agent International Limited Allergan Sales, LLC Clonshaugh Industrial Estate 2525 Dupont Drive Coolock Irvine, California USA 92612 Dublin 17 +1-714-246-4500 Ireland +1-800-347-4500 Emergency Telephone Number(s): Refer to the Study Contacts Page Serious Adverse Event Reporting Fax Number: Allergan Medical Safety Physician Please see Study Contact Sheet Contact Information: Allergan Signatory: Original Protocol Date: 02 May 2016 Protocol Amendment 1 Date: 15 Nov 2016 Protocol Amendment 2 Date: 06 Dec 2017 Protocol Amendment 3 Date: 11 Apr 2018 The following information can be found on FDA Form 1572 and/or study contacts page: Name and contact information of Allergan study personnel and Emergency Telephone Numbers; name, address, and statement of qualifications of each investigator; name of each subinvestigator working under the supervision of the investigator; name and address of the research facilities to be used; name and address of each reviewing IRB; US 21 CFR 312.23 section 6(iii)b. -

Aimovig, INN-Erenumab
Part VI: Summary of the risk management plan Summary of risk management plan for Aimovig (Erenumab) This is a summary of the risk management plan (RMP) for Aimovig. The RMP details important risks of Aimovig, how these risks can be minimized, and how more information will be obtained about Aimovig’s risks and uncertainties (missing information). Aimovig’s summary of product characteristics (SmPC) and its package leaflet give essential information to healthcare professionals and patients on how Aimovig should be used. This summary of the RMP for Aimovig should be read in the context of all this information including the assessment report of the evaluation and its plain-language summary, all which is part of the European Public Assessment Report (EPAR). Important new concerns or changes to the current ones will be included in updates of Aimovig’s RMP. I. The medicine and what it is used for Aimovig is authorized for the prophylaxis of migraine in adults who have at least 4 migraine days per month (see SmPC for the full indication). It contains erenumab (a human IgG2 monoclonal antibody) as the active substance and it is given by sc injections. Further information about the evaluation of Aimovig’s benefits can be found in Aimovig’s EPAR, including in its plain-language summary, available on the EMA website, under the medicine’s webpage: http://www.ema.europa.eu/ema/index.jsp?curl=/pages/medicines/human/medicines/004447/hum an_med_002275.jsp. II. Risks associated with the medicine and activities to minimize or further characterize the risks Important risks of Aimovig, together with measures to minimize such risks and the proposed studies for learning more about Aimovig's risks, are outlined below in Table 2. -

Comparative Efficacy and Safety of Rimegepant Versus Ubrogepant and Lasmiditan for Acute Poster No
Comparative Efficacy and Safety of Rimegepant Versus Ubrogepant and Lasmiditan for Acute Poster No. P6.002 Treatment of Migraine: A Network Meta-analysis (NMA) 1 1 1 1 2 2 2 2 2 Karissa Johnston, PhD ; Evan Popoff, MSc ; Alison Deighton, BASc ; Parisa Dabirvaziri, MD ; Linda Harris, MPH ; Alexandra Thiry, PhD ; Robert Croop, MD ; Vladimir Coric, MD ; Gilbert L’Italien, PhD 1 Broadstreet Health Economics & Outcomes Research, Vancouver, BC; 2 Biohaven Pharmaceuticals, New Haven, CT Introduction Methods cont. Figure 3. Network Meta-analysis Results for Efficacy (Rimegepant vs Comparators) • Triptans are the current standard of care for the acute treatment of moderate to severe migraine attacks • Efficacy outcomes included sustained pain freedom and sustained pain relief 2-24 hours post-dose • Despite a demonstrated clinical benefit, response to treatment decreases over time, and triptans are • Safety outcomes included somnolence, dizziness, and nausea contraindicated in patients with cardiovascular conditions1 • Results are expressed in terms of risk difference — the incremental proportion of respondents achieving the • Novel agents for acute migraine treatment include the: outcome of interest across treatments – Calcitonin gene-related peptide (CGRP)-receptor agonists rimegepant orally disintegrating tablet (ODT) and ubrogepant oral tablet Results – 5-HT1F receptor agonist lasmiditan • Five randomized placebo-controlled trials contributed data to the network: • The safety and efficacy of these treatments have been investigated independently -

761077Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 761077Orig1s000 OTHER REVIEW(S) Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research | Office of Surveillance and Epidemiology (OSE) Epidemiology: ARIA Sufficiency Templates Version: 2018-01-24 Date: May 15, 2018 Reviewer(s): Hongliu Ding, MD, PhD, MPH Division of Epidemiology I Team Leader: Kira Leishear, PhD, MS Division of Epidemiology I Division Deputy Director: Sukhminder K. Sandhu, PhD, MS, MPH Division of Epidemiology I Subject: ARIA Sufficiency Memo for Pregnancy Safety Concerns Drug Name(s): Amovig (erenumab) Application Type/Number: BLA 761077 Applicant/sponsor: Amgen Inc. OSE RCM #: 2018-869 Page 1 of 5 Reference ID: 4263424 Expedited ARIA Sufficiency Template for Pregnancy Safety Concerns 1. BACKGROUND INFORMATION 1.1. Medical Product Amovig (erenumab) is a human immunoglobulin G2 (IgG2) monoclonal antibody against the calcitonin gene-related peptide (CGRP) receptor. CGRP is a neuropeptide that modulates nociceptive signaling and a vasodilator associated with migraine pathophysiology.1-3 Plasma CGRP levels have been shown to increase significantly during migraine and return to normal when headache is relieved.4, 5 Erenumab competes with the binding of CGRP and inhibits its function at the CGRP receptor, and thus, the proposed indication is for prophylaxis of episodic and chronic migraine in adults. 1.2. Describe the Safety Concern Amovig (erenumab) exposure to women affected by migraine that are pregnant or of childbearing potential is possible. The data from a non-clinical study provided by the sponsor in which female monkeys were administered erenumab (0 or 50 mg/kg) twice weekly by subcutaneous showed that injection throughout pregnancy (gestation day 20-22 to parturition) did not observe adverse effects on offspring. -

Preferred Drug List
Kansas State Employee ANALGESICS Second Generation cefprozil Health Plan NSAIDs cefuroxime axetil diclofenac sodium delayed-rel Preferred Drug List diflunisal Third Generation etodolac cefdinir 2021 ibuprofen cefixime (SUPRAX) meloxicam nabumetone Erythromycins/Macrolides naproxen sodium tabs azithromycin naproxen tabs clarithromycin oxaprozin clarithromycin ext-rel sulindac erythromycin delayed-rel erythromycin ethylsuccinate NSAIDs, COMBINATIONS erythromycin stearate diclofenac sodium delayed-rel/misoprostol fidaxomicin (DIFICID) Effective 04/01/2021 NSAIDs, TOPICAL Fluoroquinolones diclofenac sodium gel 1% ciprofloxacin For questions or additional information, diclofenac sodium soln levofloxacin access the State of Kansas website at moxifloxacin http://www.kdheks.gov/hcf/sehp or call COX-2 INHIBITORS Penicillins the Kansas State Employees Prescription celecoxib amoxicillin Drug Program at 1-800-294-6324. amoxicillin/clavulanate The Preferred Drug List is subject to change. GOUT amoxicillin/clavulanate ext-rel To locate covered prescriptions online, allopurinol ampicillin access the State of Kansas website at colchicine tabs dicloxacillin http://www.kdheks.gov/hcf/sehp for the probenecid penicillin VK most current drug list. colchicine (MITIGARE) Tetracyclines What is a Preferred Drug List? OPIOID ANALGESICS doxycycline hyclate A Preferred Drug List is a list of safe and buprenorphine transdermal minocycline cost-effective drugs, chosen by a committee codeine/acetaminophen tetracycline of physicians and pharmacists. Drug lists fentanyl -

CDER List of Licensed Biological Products With
Center for Drug Evaluation and Research List of Licensed Biological Products with (1) Reference Product Exclusivity and (2) Biosimilarity or Interchangeability Evaluations to Date DATE OF FIRST REFERENCE PRODUCT DATE OF LICENSURE LICENSURE EXCLUSIVITY EXPIRY DATE INTERCHANGEABLE (I)/ BLA STN PRODUCT (PROPER) NAME PROPRIETARY NAME (mo/day/yr) (mo/day/yr) (mo/day/yr) BIOSIMILAR (B) WITHDRAWN 125118 abatacept Orencia 12/23/05 NA NA 103575 abciximab ReoPro 12/22/94 NA NA Yes 125274 abobotulinumtoxinA Dysport 04/29/09 125057 adalimumab Humira 12/31/02 NA NA 761071 adalimumab-adaz Hyrimoz 10/30/18 B 761058 adalimumab-adbm Cyltezo 08/25/17 B 761118 adalimumab-afzb Abrilada 11/15/19 B 761024 adalimumab-atto Amjevita 09/23/16 B 761059 adalimumab-bwwd Hadlima 07/23/19 B 125427 ado-trastuzumab emtansine Kadcyla 02/22/13 125387 aflibercept Eylea 11/18/11 103979 agalsidase beta Fabrazyme 04/24/03 NA NA 125431 albiglutide Tanzeum 04/15/14 017835 albumin chromated CR-51 serum Chromalbin 02/23/76 103293 aldesleukin Proleukin 05/05/92 NA NA 103948 alemtuzumab Campath, Lemtrada 05/07/01 NA NA 125141 alglucosidase alfa Myozyme 04/28/06 NA NA 125291 alglucosidase alfa Lumizyme 05/24/10 125559 alirocumab Praluent 07/24/15 103172 alteplase, cathflo activase Activase 11/13/87 NA NA 103950 anakinra Kineret 11/14/01 NA NA 020304 aprotinin Trasylol 12/29/93 125513 asfotase alfa Strensiq 10/23/15 101063 asparaginase Elspar 01/10/78 NA NA 125359 asparaginase erwinia chrysanthemi Erwinaze 11/18/11 761034 atezolizumab Tecentriq 05/18/16 761049 avelumab Bavencio 03/23/17