Large-Scale Discovery of Mouse Transgenic Integration Sites Reveals Frequent Structural Variation and Insertional Mutagenesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
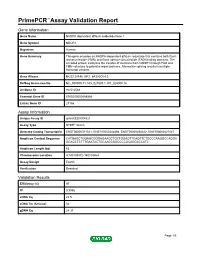
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name NADPH dependent diflavin oxidoreductase 1 Gene Symbol NDOR1 Organism Human Gene Summary This gene encodes an NADPH-dependent diflavin reductase that contains both flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) binding domains. The encoded protein catalyzes the transfer of electrons from NADPH through FAD and FMN cofactors to potential redox partners. Alternative splicing results in multiple transcript variants. Gene Aliases MGC138148, NR1, bA350O14.9 RefSeq Accession No. NC_000009.11, NG_027801.1, NT_024000.16 UniGene ID Hs.512564 Ensembl Gene ID ENSG00000188566 Entrez Gene ID 27158 Assay Information Unique Assay ID qHsaCED0004821 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000371521, ENST00000344894, ENST00000458322, ENST00000427047 Amplicon Context Sequence CATGAGCTGGAGCGGGAGAAGCTGCTGGAGTTCAGTTCTGCCCAAGGCCAGGA GGAGCTCTTTGAATACTGCAACCGGCCCCGCAGGACCATC Amplicon Length (bp) 63 Chromosome Location 9:140109372-140109464 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9996 cDNA Cq 22.5 cDNA Tm (Celsius) 82 gDNA Cq 24.37 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report NDOR1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

UNIVERSITY of CALIFORNIA, SAN DIEGO Biochemical and Structural
UNIVERSITY OF CALIFORNIA, SAN DIEGO Biochemical and Structural Characterization of the NEET Protein Family and their Role in Iron Homeostasis and Disease A Dissertation submitted in partial satisfaction of the requirements of the degree Doctor of Philosophy in Chemistry by Colin Harrison Lipper Committee in charge: Professor Patricia A. Jennings, Chair Professor Joseph A. Adams Professor Michael Galperin Professor Stanley J. Opella Professor Navtej Toor 2017 Copyright Colin Harrison Lipper, 2017 All rights reserved The dissertation of Colin Harrison Lipper is approved, and it is acceptable in quality and form for publication on microfilm and electronically: Chair UNIVERSITY OF CALIFORNIA, SAN DIEGO 2017 iii DEDICATION To my wife Hope. Thank you for all of your love and support. I could not have done this without you. iv TABLE OF CONTENTS Signature Page ................................................................................................ iii Dedication ....................................................................................................... iv Table of Contents ............................................................................................. v List of Figures .................................................................................................. vi List of Tables ................................................................................................... ix List of Abbreviations ..............................................................................……... x Acknowledgements ....................................................................................... -

Role of GSH and Iron-Sulfur Glutaredoxins in Iron Metabolism—Review
molecules Review Role of GSH and Iron-Sulfur Glutaredoxins in Iron Metabolism—Review 1, 1, 1 1 Trnka Daniel y , Hossain Md Faruq y , Jordt Laura Magdalena , Gellert Manuela and Lillig Christopher Horst 2,* 1 Institute for Medical Biochemistry and Molecular Biology, University Medicine, University of Greifswald, 17475 Greifswald, Germany; [email protected] (T.D.); [email protected] (H.M.F.); [email protected] (J.L.M.); [email protected] (G.M.) 2 Christopher Horst Lillig, Institute for Medical Biochemistry and Molecular Biology, University Medicine Greifswald, Ferdinand-Sauerbruch-Straße, 17475 Greifswald, Germany * Correspondence: [email protected]; Tel.: +49-3834-865407; Fax: +49-3834-865402 These authors contributed equally to this work. y Academic Editor: Pál Perjési Received: 29 July 2020; Accepted: 22 August 2020; Published: 25 August 2020 Abstract: Glutathione (GSH) was initially identified and characterized for its redox properties and later for its contributions to detoxification reactions. Over the past decade, however, the essential contributions of glutathione to cellular iron metabolism have come more and more into focus. GSH is indispensable in mitochondrial iron-sulfur (FeS) cluster biosynthesis, primarily by co-ligating FeS clusters as a cofactor of the CGFS-type (class II) glutaredoxins (Grxs). GSH is required for the export of the yet to be defined FeS precursor from the mitochondria to the cytosol. In the cytosol, it is an essential cofactor, again of the multi-domain CGFS-type Grxs, master players in cellular iron and FeS trafficking. In this review, we summarize the recent advances and progress in this field. The most urgent open questions are discussed, such as the role of GSH in the export of FeS precursors from mitochondria, the physiological roles of the CGFS-type Grx interactions with BolA-like proteins and the cluster transfer between Grxs and recipient proteins. -

Genomic and Transcriptomic Investigations Into the Feed Efficiency Phenotype of Beef Cattle
Provided by the author(s) and NUI Galway in accordance with publisher policies. Please cite the published version when available. Title Genomic and transcriptomic investigations into the feed efficiency phenotype of beef cattle Author(s) Higgins, Marc Publication Date 2019-03-06 Publisher NUI Galway Item record http://hdl.handle.net/10379/15008 Downloaded 2021-09-25T18:07:39Z Some rights reserved. For more information, please see the item record link above. Genomic and Transcriptomic Investigations into the Feed Efficiency Phenotype of Beef Cattle Marc Higgins, B.Sc., M.Sc. A thesis submitted for the Degree of Doctor of Philosophy to the Discipline of Biochemistry, School of Natural Sciences, National University of Ireland, Galway. Supervisor: Dr. Derek Morris Discipline of Biochemistry, School of Natural Sciences, National University of Ireland, Galway. Supervisor: Dr. Sinéad Waters Teagasc, Animal and Bioscience Research Department, Animal & Grassland Research and Innovation Centre, Teagasc, Grange. Submitted November 2018 Table of Contents Declaration ................................................................................................................ vii Funding .................................................................................................................... viii Acknowledgements .................................................................................................... ix Abstract ...................................................................................................................... -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Fe-S Protein Synthesis in Green Algae Mitochondria
plants Review Fe-S Protein Synthesis in Green Algae Mitochondria Diego F. Gomez-Casati * , Maria V. Busi *, Julieta Barchiesi, Maria A. Pagani , Noelia S. Marchetti-Acosta and Agustina Terenzi Centro de Estudios Fotosintéticos y Bioquímicos (CEFOBI-CONICET), Universidad Nacional de Rosario, 2000 Rosario, Argentina; [email protected] (J.B.); [email protected] (M.A.P.); [email protected] (N.S.M.-A.); [email protected] (A.T.) * Correspondence: [email protected] (D.F.G.-C.); [email protected] (M.V.B.); Tel.: +54-341-4391955 (ext. 113) (D.F.G.-C. & M.V.B.) Abstract: Iron and sulfur are two essential elements for all organisms. These elements form the Fe-S clusters that are present as cofactors in numerous proteins and protein complexes related to key processes in cells, such as respiration and photosynthesis, and participate in numerous enzymatic reactions. In photosynthetic organisms, the ISC and SUF Fe-S cluster synthesis pathways are located in organelles, mitochondria, and chloroplasts, respectively. There is also a third biosynthetic machinery in the cytosol (CIA) that is dependent on the mitochondria for its function. The genes and proteins that participate in these assembly pathways have been described mainly in bacteria, yeasts, humans, and recently in higher plants. However, little is known about the proteins that participate in these processes in algae. This review work is mainly focused on releasing the information on the existence of genes and proteins of green algae (chlorophytes) that could participate in the assembly process of Fe-S groups, especially in the mitochondrial ISC and CIA pathways. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Supporting Information
Supporting information Banci et al. 10.1073/pnas.1302378110 SI Materials and Methods Electrospray Ionization-MS, Circular Dichroism, and EPR. Electrospray Cloning and Protein Production of the FMN-Binding Domain of NADPH- ionization (ESI)-MS spectra were recorded by direct introduction dependent diflavin oxidoreductase 1. The FMN-binding domain of of the samples at 5 μL/min flow rate in an LTQ-Orbitrap high- NADPH-dependent diflavin oxidoreductase 1 (Ndor1), encoding resolution mass spectrometer (Thermo), equipped with a con- residues 1–161 or 1–174, was PCR amplified from a commercially ventional ESI source. For acquisition, Xcalibur 2.0. software acquired plasmid (IMAGE Full Length cDNA clone IRAT- (Thermo) was used and monoisotopic and average deconvo- p970E05127D) encoding human Ndor1 and inserted in Gateway luted masses were obtained by using the integrated Xtract tool. pEntr-TEV-d-Topo vector. The protein constructs were expressed For spectra acquisition a nominal resolution (at m/z 400) of with an N-terminal protein G β1 domain (GB1)-His-tag in BL21 100,000 was used. – (DE3)pLysS cells. The cultures were grown at 37 °C overnight in Circular dichroism spectra (290 340 nm) were run at 298 K with LB or minimal medium containing 100 μg/mL ampicillin, 34 μg/mL a JASCO Jasco-810 spectropolarimeter, using a 1-cm cell and chloramphenicol, and 100 μMriboflavin and grown further at 37 °C bandwidth of 1 nm. Spectra were accumulated 10 times and the values were corrected for buffer contributions. The CDPro soft- until OD600 reached 0.6–0.8. The culture was transferred to 17 °C and protein expression was induced by addition of isopropyl-β-D- ware package was used to determine the secondary structure thiogalactopyranosid (IPTG) to a final concentration of 0.5 mM content, considering reference dataset 1 (http://lamar.colostate. -

Characterizing Genomic Duplication in Autism Spectrum Disorder by Edward James Higginbotham a Thesis Submitted in Conformity
Characterizing Genomic Duplication in Autism Spectrum Disorder by Edward James Higginbotham A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Molecular Genetics University of Toronto © Copyright by Edward James Higginbotham 2020 i Abstract Characterizing Genomic Duplication in Autism Spectrum Disorder Edward James Higginbotham Master of Science Graduate Department of Molecular Genetics University of Toronto 2020 Duplication, the gain of additional copies of genomic material relative to its ancestral diploid state is yet to achieve full appreciation for its role in human traits and disease. Challenges include accurately genotyping, annotating, and characterizing the properties of duplications, and resolving duplication mechanisms. Whole genome sequencing, in principle, should enable accurate detection of duplications in a single experiment. This thesis makes use of the technology to catalogue disease relevant duplications in the genomes of 2,739 individuals with Autism Spectrum Disorder (ASD) who enrolled in the Autism Speaks MSSNG Project. Fine-mapping the breakpoint junctions of 259 ASD-relevant duplications identified 34 (13.1%) variants with complex genomic structures as well as tandem (193/259, 74.5%) and NAHR- mediated (6/259, 2.3%) duplications. As whole genome sequencing-based studies expand in scale and reach, a continued focus on generating high-quality, standardized duplication data will be prerequisite to addressing their associated biological mechanisms. ii Acknowledgements I thank Dr. Stephen Scherer for his leadership par excellence, his generosity, and for giving me a chance. I am grateful for his investment and the opportunities afforded me, from which I have learned and benefited. I would next thank Drs. -

Neuroplasticity Pathways and Protein-Interaction Networks Are
Waller et al. BMC Neurosci (2017) 18:56 DOI 10.1186/s12868-017-0376-x BMC Neuroscience RESEARCH ARTICLE Open Access Neuroplasticity pathways and protein‑interaction networks are modulated by vortioxetine in rodents Jessica A. Waller1, Sara Holm Nygaard2, Yan Li1, Kristian Gaarn du Jardin3, Joseph A. Tamm4, Aicha Abdourahman4, Betina Elfving3, Alan L. Pehrson1, Connie Sánchez1* and Rasmus Wernersson2,5* Abstract Background: The identifcation of biomarkers that predict susceptibility to major depressive disorder and treatment response to antidepressants is a major challenge. Vortioxetine is a novel multimodal antidepressant that possesses pro-cognitive properties and diferentiates from other conventional antidepressants on various cognitive and plastic- ity measures. The aim of the present study was to identify biological systems rather than single biomarkers that may underlie vortioxetine’s treatment efects. Results: We show that the biological systems regulated by vortioxetine are overlapping between mouse and rat in response to distinct treatment regimens and in diferent brain regions. Furthermore, analysis of complexes of physically-interacting proteins reveal that biomarkers involved in transcriptional regulation, neurodevelopment, neu- roplasticity, and endocytosis are modulated by vortioxetine. A subsequent qPCR study examining the expression of targets in the protein–protein interactome space in response to chronic vortioxetine treatment over a range of doses provides further biological validation that vortioxetine engages neuroplasticity networks. Thus, the same biology is regulated in diferent species and sexes, diferent brain regions, and in response to distinct routes of administration and regimens. Conclusions: A recurring theme, based on the present study as well as previous fndings, is that networks related to synaptic plasticity, synaptic transmission, signal transduction, and neurodevelopment are modulated in response to vortioxetine treatment.