Genetic Algorithm-Neural Network: Feature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Efficacy and Mechanistic Evaluation of Tic10, a Novel Antitumor Agent
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2012 Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Joshua Edward Allen University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Oncology Commons Recommended Citation Allen, Joshua Edward, "Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent" (2012). Publicly Accessible Penn Dissertations. 488. https://repository.upenn.edu/edissertations/488 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/488 For more information, please contact [email protected]. Efficacy and Mechanisticv E aluation of Tic10, A Novel Antitumor Agent Abstract TNF-related apoptosis-inducing ligand (TRAIL; Apo2L) is an endogenous protein that selectively induces apoptosis in cancer cells and is a critical effector in the immune surveillance of cancer. Recombinant TRAIL and TRAIL-agonist antibodies are in clinical trials for the treatment of solid malignancies due to the cancer-specific cytotoxicity of TRAIL. Recombinant TRAIL has a short serum half-life and both recombinant TRAIL and TRAIL receptor agonist antibodies have a limited capacity to perfuse to tissue compartments such as the brain, limiting their efficacy in certain malignancies. To overcome such limitations, we searched for small molecules capable of inducing the TRAIL gene using a high throughput luciferase reporter gene assay. We selected TRAIL-inducing compound 10 (TIC10) for further study based on its induction of TRAIL at the cell surface and its promising therapeutic index. TIC10 is a potent, stable, and orally active antitumor agent that crosses the blood-brain barrier and transcriptionally induces TRAIL and TRAIL-mediated cell death in a p53-independent manner. -

Supplementary Data
Supplementary Fig. 1 A B Responder_Xenograft_ Responder_Xenograft_ NON- NON- Lu7336, Vehicle vs Lu7466, Vehicle vs Responder_Xenograft_ Responder_Xenograft_ Sagopilone, Welch- Sagopilone, Welch- Lu7187, Vehicle vs Lu7406, Vehicle vs Test: 638 Test: 600 Sagopilone, Welch- Sagopilone, Welch- Test: 468 Test: 482 Responder_Xenograft_ NON- Lu7860, Vehicle vs Responder_Xenograft_ Sagopilone, Welch - Lu7558, Vehicle vs Test: 605 Sagopilone, Welch- Test: 333 Supplementary Fig. 2 Supplementary Fig. 3 Supplementary Figure S1. Venn diagrams comparing probe sets regulated by Sagopilone treatment (10mg/kg for 24h) between individual models (Welsh Test ellipse p-value<0.001 or 5-fold change). A Sagopilone responder models, B Sagopilone non-responder models. Supplementary Figure S2. Pathway analysis of genes regulated by Sagopilone treatment in responder xenograft models 24h after Sagopilone treatment by GeneGo Metacore; the most significant pathway map representing cell cycle/spindle assembly and chromosome separation is shown, genes upregulated by Sagopilone treatment are marked with red thermometers. Supplementary Figure S3. GeneGo Metacore pathway analysis of genes differentially expressed between Sagopilone Responder and Non-Responder models displaying –log(p-Values) of most significant pathway maps. Supplementary Tables Supplementary Table 1. Response and activity in 22 non-small-cell lung cancer (NSCLC) xenograft models after treatment with Sagopilone and other cytotoxic agents commonly used in the management of NSCLC Tumor Model Response type -

Supplemental Figure S1 Differentially Methylated Regions (Dmrs
Supplemental Figure S1 '$$#0#,2'**7+#2&7*2#"0#%'-,11 #25##,"'1#1#122#1 '!2-0'*"#.'!2'-,-$122,1'2'-,$0-+2- !"Q !"2-$%," $ 31',% 25-$-*" !&,%# ," ' 0RTRW 1 !32V-$$ !0'2#0'T - #.0#1#,22'-, -$ "'$$#0#,2'**7+#2&7*2#"%#,#11',.0#,2#1,"2&#'0 #&4'-022,1'2'-, #25##,"'$$#0#,2"'1#1#122#1T-*!)00-51',"'!2#&7.#0+#2&7*2#"%#,#1Q%0700-51 &7.-+#2&7*2#"%#,#1Q31',%25-$-*"!&,%#,"'0RTRW1!32V-$$!0'2#0'T-%#,#1 +#22&# -4#!0'2#0'22,1'2'-,$0-+$%2-$Q5#2�#$-0#*1-',!*3"#" %#,#15'2&V4*3#0RTRWT$$#!2#"%#,10#&'%&*'%&2#" 712#0'1)1#T Supplemental Figure S2 Validation of results from the HELP assay using Epityper MassarrayT #13*21 $0-+ 2&# 1$ 117 5#0# !-00#*2#" 5'2& /3,2'22'4# +#2&7*2'-, ,*78#" 7 '13*$'2#11007$-04V-,"6U-%#,#.0-+-2#00#%'-,1T11007 51.#0$-0+#"31',%**4'* *#1+.*#1T S Supplemental Fig. S1 A unique hypermethylated genes (methylation sites) 454 (481) 5693 (6747) 120 (122) NLMGUS NEWMM REL 2963 (3207) 1338 (1560) 5 (5) unique hypomethylated genes (methylation sites) B NEWMM 0 (0) MGUS 454 (481) 0 (0) NEWMM REL NL 3* (2) 2472 (3066) NEWMM 2963 REL (3207) 2* (2) MGUS 0 (0) REL 2 (2) NEWMM 0 (0) REL Supplemental Fig. S2 A B ARID4B DNMT3A Methylation by MassArray Methylation by MassArray 0 0.2 0.4 0.6 0.8 1 1.2 0.5 0.6 0.7 0.8 0.9 1 2 0 NL PC MGUS 1.5 -0.5 NEW MM 1 REL MM -1 0.5 -1.5 0 -2 -0.5 -1 -2.5 -1.5 -3 Methylation by HELP Assay Methylation by HELP Methylation by HELP Assay Methylation by HELP -2 -3.5 -2.5 -4 Supplemental tables "3..*#+#,2*6 *#"SS 9*','!*!&0!2#0'12'!1-$.2'#,21+.*#1 DZ_STAGE Age Gender Ethnicity MM isotype PCLI Cytogenetics -

Genome-Wide Transcriptome Analysis of Laminar Tissue During the Early Stages of Experimentally Induced Equine Laminitis
GENOME-WIDE TRANSCRIPTOME ANALYSIS OF LAMINAR TISSUE DURING THE EARLY STAGES OF EXPERIMENTALLY INDUCED EQUINE LAMINITIS A Dissertation by JIXIN WANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY December 2010 Major Subject: Biomedical Sciences GENOME-WIDE TRANSCRIPTOME ANALYSIS OF LAMINAR TISSUE DURING THE EARLY STAGES OF EXPERIMENTALLY INDUCED EQUINE LAMINITIS A Dissertation by JIXIN WANG Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Approved by: Chair of Committee, Bhanu P. Chowdhary Committee Members, Terje Raudsepp Paul B. Samollow Loren C. Skow Penny K. Riggs Head of Department, Evelyn Tiffany-Castiglioni December 2010 Major Subject: Biomedical Sciences iii ABSTRACT Genome-wide Transcriptome Analysis of Laminar Tissue During the Early Stages of Experimentally Induced Equine Laminitis. (December 2010) Jixin Wang, B.S., Tarim University of Agricultural Reclamation; M.S., South China Agricultural University; M.S., Texas A&M University Chair of Advisory Committee: Dr. Bhanu P. Chowdhary Equine laminitis is a debilitating disease that causes extreme sufferring in afflicted horses and often results in a lifetime of chronic pain. The exact sequence of pathophysiological events culminating in laminitis has not yet been characterized, and this is reflected in the lack of any consistently effective therapeutic strategy. For these reasons, we used a newly developed 21,000 element equine-specific whole-genome oligoarray to perform transcriptomic analysis on laminar tissue from horses with experimentally induced models of laminitis: carbohydrate overload (CHO), hyperinsulinaemia (HI), and oligofructose (OF). -

A Systems Approach to Prion Disease
Molecular Systems Biology 5; Article number 252; doi:10.1038/msb.2009.10 Citation: Molecular Systems Biology 5:252 & 2009 EMBO and Macmillan Publishers Limited All rights reserved 1744-4292/09 www.molecularsystemsbiology.com A systems approach to prion disease Daehee Hwang1,2,8, Inyoul Y Lee1,8, Hyuntae Yoo1,8, Nils Gehlenborg1,3, Ji-Hoon Cho2, Brianne Petritis1, David Baxter1, Rose Pitstick4, Rebecca Young4, Doug Spicer4, Nathan D Price7, John G Hohmann5, Stephen J DeArmond6, George A Carlson4,* and Leroy E Hood1,* 1 Institute for Systems Biology, Seattle, WA, USA, 2 I-Bio Program & Department of Chemical Engineering, POSTECH, Pohang, Republic of Korea, 3 Microarray Team, European Bioinformatics Institute, Wellcome Trust Genome Campus, Cambridge, UK, 4 McLaughlin Research Institute, Great Falls, MT, USA, 5 Allen Brain Institute, Seattle, WA, USA, 6 Department of Pathology, University of California, San Francisco, CA, USA and 7 Department of Chemical and Biomolecular Engineering & Institute for Genomic Biology, University of Illinois, Urbana, IL, USA 8 These authors contributed equally to this work * Corresponding authors. GA Carlson, McLaughlin Research Institute, 1520 23rd Street South, Great Falls, MT 59405, USA. Tel.: þ 1 406 454 6044; Fax: þ 1 406 454 6019; E-mail: [email protected] or LE Hood, Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103, USA. Tel.: þ 1 206 732 1201; Fax: þ 1 206 732 1254; E-mail: [email protected] Received 27.11.08; accepted 20.1.09 Prions cause transmissible neurodegenerative diseases and replicate by conformational conversion of normal benign forms of prion protein (PrPC) to disease-causing PrPSc isoforms. -
Rat Mitochondrion-Neuron Focused Microarray (Rmnchip) and Bioinfor- Matics Tools for Rapid Identification of Differential Pathways in Brain Tissues Yan A
Int. J. Biol. Sci. 2011, 7 308 International Journal of Biological Sciences 2011; 7(3):308-322 Research Paper Rat Mitochondrion-Neuron Focused Microarray (rMNChip) and Bioinfor- matics Tools for Rapid Identification of Differential Pathways in Brain Tissues Yan A. Su, Qiuyang Zhang, David M. Su, Michael X. Tang Department of Gene and Protein Biomarkers, GenProMarkers Inc., Rockville, MD 20850, USA Corresponding author: Yan A. Su, M.D., Ph.D., GenProMarkers Inc., 9700 Great Seneca Highway, Suite 182, Rockville, Maryland 20850. Phone: (301) 326-6523; Email:[email protected] © Ivyspring International Publisher. This is an open-access article distributed under the terms of the Creative Commons License (http://creativecommons.org/ licenses/by-nc-nd/3.0/). Reproduction is permitted for personal, noncommercial use, provided that the article is in whole, unmodified, and properly cited. Received: 2011.01.01; Accepted: 2011.03.25; Published: 2011.03.29 Abstract Mitochondrial function is of particular importance in brain because of its high demand for energy (ATP) and efficient removal of reactive oxygen species (ROS). We developed rat mitochondrion-neuron focused microarray (rMNChip) and integrated bioinformatics tools for rapid identification of differential pathways in brain tissues. rMNChip contains 1,500 genes involved in mitochondrial functions, stress response, circadian rhythms and signal transduc- tion. The bioinformatics tool includes an algorithm for computing of differentially expressed genes, and a database for straightforward -

Transdifferentiation of Human Mesenchymal Stem Cells
Transdifferentiation of Human Mesenchymal Stem Cells Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Julius-Maximilians-Universität Würzburg vorgelegt von Tatjana Schilling aus San Miguel de Tucuman, Argentinien Würzburg, 2007 Eingereicht am: Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Martin J. Müller Gutachter: PD Dr. Norbert Schütze Gutachter: Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am: Hiermit erkläre ich ehrenwörtlich, dass ich die vorliegende Dissertation selbstständig angefertigt und keine anderen als die von mir angegebenen Hilfsmittel und Quellen verwendet habe. Des Weiteren erkläre ich, dass diese Arbeit weder in gleicher noch in ähnlicher Form in einem Prüfungsverfahren vorgelegen hat und ich noch keinen Promotionsversuch unternommen habe. Gerbrunn, 4. Mai 2007 Tatjana Schilling Table of contents i Table of contents 1 Summary ........................................................................................................................ 1 1.1 Summary.................................................................................................................... 1 1.2 Zusammenfassung..................................................................................................... 2 2 Introduction.................................................................................................................... 4 2.1 Osteoporosis and the fatty degeneration of the bone marrow..................................... 4 2.2 Adipose and bone -

Encoded on Chromosome 6P21.33 in Human Breast Cancers Revealed by Transcrip- Tome Analysis Yan A
Journal of Cancer 2010, 1 38 Journal of Cancer 2010; 1:38-50 © Ivyspring International Publisher. All rights reserved Research Paper Undetectable and Decreased Expression of KIAA1949 (Phostensin) Encoded on Chromosome 6p21.33 in Human Breast Cancers Revealed by Transcrip- tome Analysis Yan A. Su1 , Jun Yang1, Lian Tao1, and Hein Nguyen1, and Ping He2 1. GenProMarkers Inc., Rockville, Maryland 20850, USA; 2. Division of Hematology, Center for Biological Evaluation and Research, Food and Drug Administration, Bethesda, MD 20892, USA Corresponding author: Yan A. Su, MD, PhD, GenProMarkers Inc., 9700 Great Seneca Highway, Suite 182, Rockville, Maryland 20850. Phone: (301) 326-6523; FAX: (240) 453-6208; Email:[email protected] Published: 2010.06.21 Abstract Cytogenetic aberration and loss of heterozygosity (LOH) are documented on chromosome 6 in many cancers and the introduction of a neo-tagged chromosome 6 into breast cancer cell lines mediates suppression of tumorigenicity. In this study, we described the identification of KIAA1949 (phostensin) as a putative tumor suppressor gene. Our microarray analysis screened 25,985 cDNAs between a tumorigenic and metastatic breast cancer cell line MDA-MB-231 and the chromosome 6-mediated suppressed, non-tumorigenic and non-metastatic derivative cell line MDA/H6, resulting in the identification of 651 differentially expressed genes. Using customized microarrays containing these 651 cDNAs and 117 con- trols, we identified 200 frequently dysregulated genes in 10 breast cancer cell lines and 5 tumor tissues using MDA/H6 as reference. Our bioinformatics analysis revealed that chro- mosome 6 encodes 25 of these 200 genes, with 4 downregulation and 21 upergulation. -
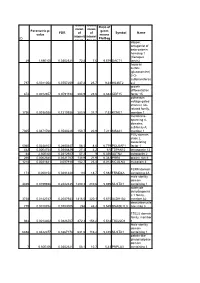
ID Parametric P
Geom Geom Ratio of mean mean Parametric p- geom FDR of of Symbol Name value means intensit intensi ID Pla/Bag ies in ties in dapper, antagonist of beta-catenin, homolog 1 (Xenopus 29 1,69E-05 0,0302321 72,8 7,6 9,579 DACT1 laevis) heparan sulfate (glucosamine) 3-O- sulfotransferas 797 0,0011054 0,0757259 237,2 25,7 9,23 HS3ST2 e 2 growth differentiation 874 0,0012657 0,0791194 204,9 23,6 8,682 GDF15 factor 15 potassium voltage-gated channel, Isk- related family, 1756 0,0038928 0,1210936 283,5 37,7 7,52 KCNE1 member 1 membrane- spanning 4- domains, subfamily A, 7305 0,0471766 0,3530226 150,7 20,9 7,211 MS4A1 member 1 POU domain, class 2, associating 5060 0,0248103 0,2680447 58,3 8,6 6,779 POU2AF1 factor 1 442 0,0004744 0,0586829 27,3 4,2 6,5 TSPAN12 tetraspanin 12 44 2,60E-05 0,0312927 57,2 9 6,356 ASTN2 astrotactin 2 266 0,0002545 0,0521767 138,9 21,9 6,342 PRR6 proline rich 6 1218 0,0021841 0,097918 152,7 25,4 6,012 MCOLN3 mucolipin 3 FERM domain 173 0,000154 0,0481428 110 18,7 5,882 FRMD4A containing 4A male sterility domain 4469 0,0199658 0,2442435 1200,3 210,6 5,699 MLSTD1 containing 1 aldehyde dehydrogenas e 1 family, 3738 0,0142747 0,2087933 1816,5 320,1 5,675 ALDH1A2 member A2 deoxyribonucle 779 0,0010754 0,0753005 268 48,3 5,549 DNASE1L3 ase I-like 3 TSC22 domain family, member 944 0,0014302 0,0826707 872,3 158,2 5,514 TSC22D1 1 Male sterility domain 6858 0,0422377 0,3367178 631,6 116,2 5,435 MLSTD1 containing 1 patatin-like phospholipase domain 7 6,60E-06 0,0302321 58,1 10,7 5,43 PNPLA3 containing 3 hypothetical protein 1857 -

1 SUPPLEMENTAL DATA Figure S1. Poly I:C Induces IFN-Β Expression
SUPPLEMENTAL DATA Figure S1. Poly I:C induces IFN-β expression and signaling. Fibroblasts were incubated in media with or without Poly I:C for 24 h. RNA was isolated and processed for microarray analysis. Genes showing >2-fold up- or down-regulation compared to control fibroblasts were analyzed using Ingenuity Pathway Analysis Software (Red color, up-regulation; Green color, down-regulation). The transcripts with known gene identifiers (HUGO gene symbols) were entered into the Ingenuity Pathways Knowledge Base IPA 4.0. Each gene identifier mapped in the Ingenuity Pathways Knowledge Base was termed as a focus gene, which was overlaid into a global molecular network established from the information in the Ingenuity Pathways Knowledge Base. Each network contained a maximum of 35 focus genes. 1 Figure S2. The overlap of genes regulated by Poly I:C and by IFN. Bioinformatics analysis was conducted to generate a list of 2003 genes showing >2 fold up or down- regulation in fibroblasts treated with Poly I:C for 24 h. The overlap of this gene set with the 117 skin gene IFN Core Signature comprised of datasets of skin cells stimulated by IFN (Wong et al, 2012) was generated using Microsoft Excel. 2 Symbol Description polyIC 24h IFN 24h CXCL10 chemokine (C-X-C motif) ligand 10 129 7.14 CCL5 chemokine (C-C motif) ligand 5 118 1.12 CCL5 chemokine (C-C motif) ligand 5 115 1.01 OASL 2'-5'-oligoadenylate synthetase-like 83.3 9.52 CCL8 chemokine (C-C motif) ligand 8 78.5 3.25 IDO1 indoleamine 2,3-dioxygenase 1 76.3 3.5 IFI27 interferon, alpha-inducible -

Mrna Expression in Human Leiomyoma and Eker Rats As Measured by Microarray Analysis
Table 3S: mRNA Expression in Human Leiomyoma and Eker Rats as Measured by Microarray Analysis Human_avg Rat_avg_ PENG_ Entrez. Human_ log2_ log2_ RAPAMYCIN Gene.Symbol Gene.ID Gene Description avg_tstat Human_FDR foldChange Rat_avg_tstat Rat_FDR foldChange _DN A1BG 1 alpha-1-B glycoprotein 4.982 9.52E-05 0.68 -0.8346 0.4639 -0.38 A1CF 29974 APOBEC1 complementation factor -0.08024 0.9541 -0.02 0.9141 0.421 0.10 A2BP1 54715 ataxin 2-binding protein 1 2.811 0.01093 0.65 0.07114 0.954 -0.01 A2LD1 87769 AIG2-like domain 1 -0.3033 0.8056 -0.09 -3.365 0.005704 -0.42 A2M 2 alpha-2-macroglobulin -0.8113 0.4691 -0.03 6.02 0 1.75 A4GALT 53947 alpha 1,4-galactosyltransferase 0.4383 0.7128 0.11 6.304 0 2.30 AACS 65985 acetoacetyl-CoA synthetase 0.3595 0.7664 0.03 3.534 0.00388 0.38 AADAC 13 arylacetamide deacetylase (esterase) 0.569 0.6216 0.16 0.005588 0.9968 0.00 AADAT 51166 aminoadipate aminotransferase -0.9577 0.3876 -0.11 0.8123 0.4752 0.24 AAK1 22848 AP2 associated kinase 1 -1.261 0.2505 -0.25 0.8232 0.4689 0.12 AAMP 14 angio-associated, migratory cell protein 0.873 0.4351 0.07 1.656 0.1476 0.06 AANAT 15 arylalkylamine N-acetyltransferase -0.3998 0.7394 -0.08 0.8486 0.456 0.18 AARS 16 alanyl-tRNA synthetase 5.517 0 0.34 8.616 0 0.69 AARS2 57505 alanyl-tRNA synthetase 2, mitochondrial (putative) 1.701 0.1158 0.35 0.5011 0.6622 0.07 AARSD1 80755 alanyl-tRNA synthetase domain containing 1 4.403 9.52E-05 0.52 1.279 0.2609 0.13 AASDH 132949 aminoadipate-semialdehyde dehydrogenase -0.8921 0.4247 -0.12 -2.564 0.02993 -0.32 AASDHPPT 60496 aminoadipate-semialdehyde -

Supplementary Table 1
Table S1 Control-MCI Control-AD MCI-AD ANOVA Module Membership FDR- Entrez log2 Fold log2 Fold log2 Fold corrected P- Illumina Probe ID Gene Symbol Gene Chromosome Change t P-value a Change t P-value a Change t P-value a F P-value value ILMN_2097421 MRPL51 51258 12p13.31d -0.52 -7.54 4.69E-13 -0.55 -7.28 2.40E-12 -0.04 0.02 9.83E-01 36.50 4.82E-15 8.98E-11 black ILMN_1784286 NDUFA1 4694 Xq24c -0.99 -7.35 1.56E-12 -1.08 -7.31 2.03E-12 -0.09 -0.19 8.51E-01 35.68 9.38E-15 8.98E-11 black ILMN_1815479 NOLA3 55505 15q14a -0.47 -8.11 1.04E-14 -0.23 -2.55 1.13E-02 0.25 5.42 1.14E-07 34.47 2.55E-14 1.63E-10 brown ILMN_1776104 NDUFS5 4725 1p34.3a -0.83 -6.63 1.40E-10 -0.92 -6.96 1.84E-11 -0.09 -0.55 5.84E-01 30.70 5.93E-13 2.84E-09 black ILMN_1716895 RPA3 6119 7p21.3e -0.45 -7.38 1.34E-12 -0.41 -5.80 1.52E-08 0.04 1.37 1.70E-01 30.08 1.00E-12 3.60E-09 black ILMN_1815707 CALML4 91860 15q23a -0.40 -7.31 2.02E-12 -0.37 -5.89 9.74E-09 0.03 1.23 2.21E-01 29.94 1.13E-12 3.60E-09 black ILMN_2339779 ATP6V1E1 529 22q11.21a -0.42 -7.42 1.01E-12 -0.38 -5.50 7.82E-08 0.04 1.73 8.40E-02 29.56 1.56E-12 3.92E-09 black ILMN_1726603 ATP5I 521 4p16.3d -0.70 -6.96 1.88E-11 -0.70 -6.33 8.04E-10 0.00 0.42 6.73E-01 29.50 1.64E-12 3.92E-09 black ILMN_2187718 COX17 10063 3q13.33a -0.49 -6.83 4.03E-11 -0.51 -6.33 7.79E-10 -0.02 0.29 7.69E-01 28.91 2.71E-12 5.77E-09 black ILMN_1695645 CETN2 1069 Xq28e -0.27 -5.83 1.30E-08 -0.34 -7.08 8.87E-12 -0.07 -1.45 1.47E-01 28.45 4.00E-12 7.66E-09 black ILMN_2117716 SFRS17A 8227 Xp22.33d,Yp11.32a 0.21 5.82 1.43E-08 0.25 7.05 1.08E-11