Modeling the Vapor – Liquid Equilibrium and Association
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 7.1 Nitrogen Dioxide
Chapter 7.1 Nitrogen dioxide General description Many chemical species of nitrogen oxides (NOx) exist, but the air pollutant species of most interest from the point of view of human health is nitrogen dioxide (NO2). Nitrogen dioxide is soluble in water, reddish-brown in colour, and a strong oxidant. Nitrogen dioxide is an important atmospheric trace gas, not only because of its health effects but also because (a) it absorbs visible solar radiation and contributes to impaired atmospheric visibility; (b) as an absorber of visible radiation it could have a potential direct role in global climate change if its concentrations were to become high enough; (c) it is, along with nitric oxide (NO), a chief regulator of the oxidizing capacity of the free troposphere by controlling the build-up and fate of radical species, including hydroxyl radicals; and (d) it plays a critical role in determining ozone (O3) concentrations in the troposphere because the photolysis of nitrogen dioxide is the only key initiator of the photochemical formation of ozone, whether in polluted or unpolluted atmospheres (1, 2). Sources On a global scale, emissions of nitrogen oxides from natural sources far outweigh those generated by human activities. Natural sources include intrusion of stratospheric nitrogen oxides, bacterial and volcanic action, and lightning. Because natural emissions are distributed over the entire surface of the earth, however, the resulting background atmospheric concentrations are very small. The major source of anthropogenic emissions of nitrogen oxides into the atmosphere is the combustion of fossil fuels in stationary sources (heating, power generation) and in motor vehicles (internal combustion engines). -

Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11
This PDF is available from The National Academies Press at http://www.nap.edu/catalog.php?record_id=13374 Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11 ISBN Committee on Acute Exposure Guideline Levels; Committee on 978-0-309-25481-6 Toxicology; National Research Council 356 pages 6 x 9 PAPERBACK (2012) Visit the National Academies Press online and register for... Instant access to free PDF downloads of titles from the NATIONAL ACADEMY OF SCIENCES NATIONAL ACADEMY OF ENGINEERING INSTITUTE OF MEDICINE NATIONAL RESEARCH COUNCIL 10% off print titles Custom notification of new releases in your field of interest Special offers and discounts Distribution, posting, or copying of this PDF is strictly prohibited without written permission of the National Academies Press. Unless otherwise indicated, all materials in this PDF are copyrighted by the National Academy of Sciences. Request reprint permission for this book Copyright © National Academy of Sciences. All rights reserved. Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11 Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies Copyright © National Academy of Sciences. All rights reserved. Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11 THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Insti- tute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. -

Nitrogen Oxides
Pollution Prevention and Abatement Handbook WORLD BANK GROUP Effective July 1998 Nitrogen Oxides Nitrogen oxides (NOx) in the ambient air consist 1994). The United States generates about 20 mil- primarily of nitric oxide (NO) and nitrogen di- lion metric tons of nitrogen oxides per year, about oxide (NO2). These two forms of gaseous nitro- 40% of which is emitted from mobile sources. Of gen oxides are significant pollutants of the lower the 11 million to 12 million metric tons of nitrogen atmosphere. Another form, nitrous oxide (N2O), oxides that originate from stationary sources, is a greenhouse gas. At the point of discharge about 30% is the result of fuel combustion in large from man-made sources, nitric oxide, a colorless, industrial furnaces and 70% is from electric utility tasteless gas, is the predominant form of nitro- furnaces (Cooper and Alley 1986). gen oxide. Nitric oxide is readily converted to the much more harmful nitrogen dioxide by Occurrence in Air and Routes of Exposure chemical reaction with ozone present in the at- mosphere. Nitrogen dioxide is a yellowish-or- Annual mean concentrations of nitrogen dioxide ange to reddish-brown gas with a pungent, in urban areas throughout the world are in the irritating odor, and it is a strong oxidant. A por- range of 20–90 micrograms per cubic meter (µg/ tion of nitrogen dioxide in the atmosphere is con- m3). Maximum half-hour values and maximum 24- verted to nitric acid (HNO3) and ammonium hour values of nitrogen dioxide can approach 850 salts. Nitrate aerosol (acid aerosol) is removed µg/m3 and 400 µg/m3, respectively. -

WHO Guidelines for Indoor Air Quality : Selected Pollutants
WHO GUIDELINES FOR INDOOR AIR QUALITY WHO GUIDELINES FOR INDOOR AIR QUALITY: WHO GUIDELINES FOR INDOOR AIR QUALITY: This book presents WHO guidelines for the protection of pub- lic health from risks due to a number of chemicals commonly present in indoor air. The substances considered in this review, i.e. benzene, carbon monoxide, formaldehyde, naphthalene, nitrogen dioxide, polycyclic aromatic hydrocarbons (especially benzo[a]pyrene), radon, trichloroethylene and tetrachloroethyl- ene, have indoor sources, are known in respect of their hazard- ousness to health and are often found indoors in concentrations of health concern. The guidelines are targeted at public health professionals involved in preventing health risks of environmen- SELECTED CHEMICALS SELECTED tal exposures, as well as specialists and authorities involved in the design and use of buildings, indoor materials and products. POLLUTANTS They provide a scientific basis for legally enforceable standards. World Health Organization Regional Offi ce for Europe Scherfi gsvej 8, DK-2100 Copenhagen Ø, Denmark Tel.: +45 39 17 17 17. Fax: +45 39 17 18 18 E-mail: [email protected] Web site: www.euro.who.int WHO guidelines for indoor air quality: selected pollutants The WHO European Centre for Environment and Health, Bonn Office, WHO Regional Office for Europe coordinated the development of these WHO guidelines. Keywords AIR POLLUTION, INDOOR - prevention and control AIR POLLUTANTS - adverse effects ORGANIC CHEMICALS ENVIRONMENTAL EXPOSURE - adverse effects GUIDELINES ISBN 978 92 890 0213 4 Address requests for publications of the WHO Regional Office for Europe to: Publications WHO Regional Office for Europe Scherfigsvej 8 DK-2100 Copenhagen Ø, Denmark Alternatively, complete an online request form for documentation, health information, or for per- mission to quote or translate, on the Regional Office web site (http://www.euro.who.int/pubrequest). -
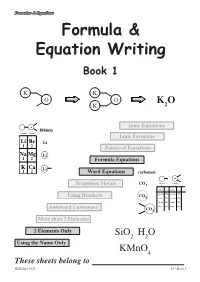
Formula & Equation Writing
Formulae & Equations Formula & Equation Writing Book 1 K K O O K O K 2 Ionic Equations lithium column column 1 2 Ionic Formulae Li Be Li 1 2 Balanced Equations Na Mg Li 1 2 Formula Equations K Ca Li 1 2 Word Equations carbonate valency valency Transition Metals CO3 1 2 name formula name formula ammonium NH4 carbonate CO3 Using Brackets CO 3 cyanide CN chromate CrO4 hydroxide OH sulphate SO4 nitrate NO sulphite SO Awkward Customers 3 3 CO3 More than 2 Elements 2 Elements Only SiO2 H2O Using the Name Only KMnO4 These sheets belong to KHS Sept 2013 page 1 S3 - Book 1 Formulae & Equations What is a Formula ? The formula of a compound tells you two things about the compound :- SiO H O i) which elements are in the compound 2 2 using symbols, and KMnO4 ii) how many atoms of each element are in the compound using numbers. Test Yourself What would be the formula for each of the following? Br H Cl K H C S Al Br O H Cl K H Br Using the Name Only Some compounds have extra information in their carbon monoxide names that allow people to work out and write CO the correct formula. carbon dioxide CO2 The names of the elements appear as usual but dinitrogen tetroxide N O this time the number of each type of atom is 2 4 included using mono- = 1 di- = 12 tri- = 3 tetra- = 4 penta- = 5 hexa- =46 Test Yourself 1 What would be the formula for each of the following? 1. -

Nitrogen Dioxide
Common Name: NITROGEN DIOXIDE CAS Number: 10102-44-0 RTK Substance number: 1376 DOT Number: UN 1067 Date: May 1989 Revision: April 2000 ----------------------------------------------------------------------- ----------------------------------------------------------------------- HAZARD SUMMARY * Nitrogen Dioxide can affect you when breathed in. * If you think you are experiencing any work-related health * Nitrogen Dioxide may cause mutations. Handle with problems, see a doctor trained to recognize occupational extreme caution. diseases. Take this Fact Sheet with you. * Contact can irritate and burn the skin and eyes with * Exposure to hazardous substances should be routinely possible eye damage. evaluated. This may include collecting personal and area * Breathing Nitrogen Dioxide can irritate the nose and air samples. You can obtain copies of sampling results throat. from your employer. You have a legal right to this * Breathing Nitrogen Dioxide can irritate the lungs causing information under OSHA 1910.1020. coughing and/or shortness of breath. Higher exposures can cause a build-up of fluid in the lungs (pulmonary edema), a medical emergency, with severe shortness of WORKPLACE EXPOSURE LIMITS breath. OSHA: The legal airborne permissible exposure limit * High levels can interfere with the ability of the blood to (PEL) is 5 ppm, not to be exceeded at any time. carry Oxygen causing headache, fatigue, dizziness, and a blue color to the skin and lips (methemoglobinemia). NIOSH: The recommended airborne exposure limit is Higher levels can cause trouble breathing, collapse and 1 ppm, which should not be exceeded at any even death. time. * Repeated exposure to high levels may lead to permanent lung damage. ACGIH: The recommended airborne exposure limit is 3 ppm averaged over an 8-hour workshift and IDENTIFICATION 5 ppm as a STEL (short term exposure limit). -

Nitric Acid Plants (PDF)
Office of Air and Radiation December 2010 AVAILABLE AND EMERGING TECHNOLOGIES FOR REDUCING GREENHOUSE GAS EMISSIONS FROM THE NITRIC ACID PRODUCTION INDUSTRY Available and Emerging Technologies for Reducing Greenhouse Gas Emissions from the Nitric Acid Production Industry Prepared by the Sector Policies and Programs Division Office of Air Quality Planning and Standards U.S. Environmental Protection Agency Research Triangle Park, North Carolina 27711 December 2010 Table of Contents Abbreviations and Acronyms p.1 I Introduction /Purpose of this Document p.2 II. Description of the Nitric Acid Production Process p.3 A. Weak Nitric Acid Production p.3 B. High-Strength Nitric Acid Production p.6 III. N2O Emissions and Nitric Acid Production Process p. 7 IV. Summary of Control Measures p.9 A. Primary Controls p.10 B. Secondary Control p.11 C. Tertiary Controls p.13 D. Selective Catalytic Reduction p.16 V. Other Greenhouse Gas Emissions p.16 VI. Energy Efficiency Improvements p.17 VII. EPA Contacts p.19 VIII. References p.20 Appendix A – Southeast Idaho Energy p.23 Appendix B - US Nitric Acid Plants p.24 Appendix C – CDM Monitoring Reports p.25 Abbreviations and Acronyms atm Pressure in atmospheres BACT Best Available Control Technologies Btu British Thermal Unit Btu/lb Btu per lb of 100% nitric acid CAR Climate Action Reserve CDM Clean Development Mechanism CHP Combined Heat and Power CO2 Carbon dioxide CO2e Carbon dioxide equivalent EU European Union GHG Greenhouse Gases H2 Hydrogen IPCC Intergovernmental Panel on Climate Change IPPC Industrial Pollution Prevention and Control JI Joint Implementation kg N2O/tonne kilograms of N2O per tonne of 100% nitric acid kg CO2e/tonne kilograms of CO2e per tonne of 100% nitric acid lb N2O/ton pounds of N2O per ton of 100% nitric acid N2 Nitrogen NH3 Ammonia NO Nitric oxide NO2 Nitrogen dioxide NOx Nitrogen oxides NSCR Nonselective Catalytic Reduction N2 O4 Nitrogen tetraoxide N2O Nitrous oxide O2 Oxygen PSD Prevention of Significant Deterioration SCR Selective Catalytic Reduction TPD Tons per day 1 I. -
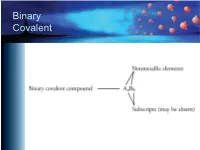
Binary Covalent Common Names
Binary Covalent Common Names – H2O, water – NH3, ammonia – CH4, methane – C2H6, ethane – C3H8, propane – C4H10, butane – C5H12, pentane – C6H14, hexane Naming Binary Covalent CompounDs • If the subscript for the first element is greater than one, inDicate the subscript with a prefix. – We Do not write mono- on the first name. – Leave the "a" off the enD of the prefixes that enD in "a" anD the “o” off of mono- if they are placeD in front of an element that begins with a vowel (oxygen or ioDine). Prefixes mon(o) hex(a) di hept(a) tri oct(a) tetr(a) non(a) pent(a) dec(a) Nitrogen OxiDe Names • N2O3 – name starts with di • N2O5 – name starts with di • NO2 – no initial prefix • NO – no initial prefix Naming Binary Covalent CompounDs • Follow the prefix with the name of the first element in the formula. – N2O3 – dinitrogen – N2O3 – dinitrogen – NO2 – nitrogen – NO – nitrogen Naming Binary Covalent CompounDs • Write a prefix to inDicate the subscript for the seconD element. (Remember to leave the “o” off of mono- anD the “a” off of the prefixes that enD in “a” when they are placeD in front of a name that begins with a vowel.) – N2O3 – dinitrogen tri – N2O5 – dinitrogen pent – NO2 – nitrogen di – NO – nitrogen mon Naming Binary Covalent CompounDs • Write the root of the name of the seconD symbol in the formula. (See the next sliDe.) – N2O3 – dinitrogen triox – N2O5 – dinitrogen pentox – NO2 – nitrogen diox – NO – nitrogen monox Roots of Nonmetals H hydr- F fluor- C carb- Cl chlor- N nitr- Br brom- P phosph- I iod- O ox- S sulf- Se selen- Naming Binary Covalent CompounDs • AdD -ide to the enD of the name. -

Supplementary Material for Zinc Oxide-Black Phosphorus Composites For
Electronic Supplementary Material (ESI) for Nanoscale Horizons. This journal is © The Royal Society of Chemistry 2018 Supplementary Material for Zinc Oxide-Black Phosphorus Composites for Ultrasensitive Nitrogen Dioxide Sensing Qun Li,ab Yuan Cen,a Jinyu Huang,a Xuejin Li,a Hao Zhang,a Youfu Geng,a Boris I. Yakobson,c Yu Du*a and Xiaoqing Tian*a aCollege of Physics and Energy, Shenzhen University, Shenzhen 518060, China. E-mail: [email protected] (Y. Du), [email protected] (X. Tian). bWuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology, Wuhan 430074, China. cDepartment of Materials Science and NanoEngineering, Department of Chemistry, Smalley Institute for Nanoscale Science and Technology, Rice University, Houston 77005, USA.* This document includes: Materials and Methods Figure S1 P 2p core-level XPS for both fresh ZnO-BP and after 24 hours, 48 hours, and 96 hours Figure S2 Response and recovery time of the ZnO-BP gas sensor to different concentrations of NO2 Figure S3 Dynamic response-recovery curves of ZnO-BP sensor to different NO2 concentrations Figure S4 Response of ZnO-BP sensor as a function of NO2 concentrations (1-10000 ppb) Figure S5 The reproducibility of the ZnO-BP sensor Figure S6 Dynamic response curves of the sensor based on ZnO-BP to 1 ppm CH3OH, CH3CH2OH, CH3COCH3, NH3, CO, SO2, and H2 Figure S7 SEM images of as-synthesized ZnO Table S1 Parameters for synthesis ZnO Materials and methods Preparation of the ZnO Hollow Spheres The aqueous solution (20 mL) of zinc acetate dehydrate (Zn(Ac)2·2H2O, 0.3512 g) and trisodium citrate dehydrate (C6H5Na3O7·2H2O, 0.12 g) were kept with mild magnetic stirring for 10 min. -

Analysis of the Relationship Between O3, NO and NO2 in Tianjin, China
Aerosol and Air Quality Research, 11: 128–139, 2011 Copyright © Taiwan Association for Aerosol Research ISSN: 1680-8584 print / 2071-1409 online doi: 10.4209/aaqr.2010.07.0055 Analysis of the Relationship between O3, NO and NO2 in Tianjin, China Suqin Han1,2, Hai Bian2, Yinchang Feng1*, Aixia Liu2, Xiangjin Li2, Fang Zeng1, Xiaoling Zhang3 1 College of Environmental Science and Engineering, Nankai University, Tianjin 300071, China 2 Tianjin Meteorological Administration, Tianjin, 300074, China 3 Beijing Meteorological Administration, Beijing, 100081, China ABSTRACT The continuous measurement of nitric oxide (NO), nitrogen dioxide (NO2), nitrogen oxides (NOx) and ozone (O3) was conducted in Tianjin from September 8 to October 15, 2006. The data were used to investigate the relationship between the O3 distribution and its association with ambient concentrations of NO, NO2 and NOx (NO and NO2). The measured concentrations of the pollutants in the study area varied as a function of time, while peaks in NO, NO2 and O3 all occurred in succession in the daytime. The diurnal cycle of ground-level ozone concentration showed a mid-day peak and lower nighttime concentrations. Furthermore, an inverse relationship was found between O3 NO, NO2 and NOx. In addition, a linear relationship between NO2 and NOx, as well as NO and NOx, and a polynomial relationship between O3 and NO2/NO was found. The variation in the level of oxidant (O3 and NO2) with NO2 was also obtained. It can be seen that OX concentration at a given location is made up of two parts: one independent and the other dependent on NO2 concentration. -

Reaction of Zn+ with NO2. the Gas-Phase Thermochemistry Of
Reaction of Zn+ with N02. The gas-phase thermochemistry of ZnO D. E. Clemmer, N. F. Dalleska, and P. B. Armentrouta) Department of Chemistry, University of Utah, Salt Lake City, Utah 84112 (Received 24 Iune 199 1; accepted 2 August 199 1) The homolytic bond dissociation energies of ZnO and ZnO + have been determined by using guided ion-beam mass spectrometry to measure the kinetic-energy dependence of the endothermic reactions of Zn + with nitrogen dioxide. The data are interpreted to yield the bond energy for ZnO, D g = 1.61 t 0.04 eV, a value considerably lower than previous experimental values, but in much better agreement with theoretical calculations. We also obtain D g (ZnO + ) = 1.67 k 0.05 eV, in good agreement with previous results. Other thermochemistry derived in this study is D i (Zn +-NO) = 0.79 f 0.10 eV and the ionization energies, IE( ZnO) = 9.34 + 0.02 eV and IE( NO, ) = 9.57 + 0.04 eV. INTRODUCTION The measurement of neutral metal-ligand thermoche- mistry from the reactions of metal ions is accomplished by In a recent paper on the thermochemistry of gas-phase studying the endothermic transfer of a negatively charged transition-metal oxide neutral and ionic diatoms,’ we noted ligand to the metal, reaction ( I ) : that there is little information about the gas-phase thermo- chemistry of zinc oxide. Zinc is the only first-row transition M+ +XL-+ML+X+ (1) metal for which there have been no definitive measurements -rML+ +X. (2) made of the neutral oxide bond energy, D O(ZnO), and ioni- zation energy, IE( ZnO). -

Nitrogen Oxides (Nox), Why and How They Are Controlled
United States Office of Air Quality EPA 456/F-99-006R Environmental Protection Planning and Standards November 1999 Agency Research Triangle Park, NC 27711 Air EPA-456/F-99-006R November 1999 Nitrogen Oxides (NOx), Why and How They Are Controlled Prepared by Clean Air Technology Center (MD-12) Information Transfer and Program Integration Division Office of Air Quality Planning and Standards U.S. Environmental Protection Agency Research Triangle Park, North Carolina 27711 DISCLAIMER This report has been reviewed by the Information Transfer and Program Integration Division of the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency and approved for publication. Approval does not signify that the contents of this report reflect the views and policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products is not intended to constitute endorsement or recommendation for use. Copies of this report are available form the National Technical Information Service, U.S. Department of Commerce, 5285 Port Royal Road, Springfield, Virginia 22161, telephone number (800) 553-6847. CORRECTION NOTICE This document, EPA-456/F-99-006a, corrects errors found in the original document, EPA-456/F-99-006. These corrections are: Page 8, fourth paragraph: “Destruction or Recovery Efficiency” has been changed to “Destruction or Removal Efficiency;” Page 10, Method 2. Reducing Residence Time: This section has been rewritten to correct for an ambiguity in the original text. Page 20, Table 4. Added Selective Non-Catalytic Reduction (SNCR) to the table and added acronyms for other technologies. Page 29, last paragraph: This paragraph has been rewritten to correct an error in stating the configuration of a typical cogeneration facility.