The Chemistry of Solvated Nitric Oxide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

United States Patent Office Patented Sept
3,149,913 United States Patent Office Patented Sept. 22, 1964 2 may vary over a wide range and may be as little as 1% 3,49,913 and as much as 50% and even higher. Particularly ad PROCESS FOR PRODUCING NETROSYL vantageous is the use of nitric acid in amounts such that SULFURECACE) d the resulting nitrosylsulfuric acid concentration approxi ALouis L. Ferstandig, El Cerrito, and Paul C. Condit, San 5 mates saturation values in order to afford maximum pro Asseino,poration, Calif.,San Francisco,assignors toCalif., California a corporation Research Cor of duction per unit reactor volume and yet avoid deposition Beavy are of solids. The solubility of nitrosylsulfuric acid in ap No Drawing. Fied June 14, 1961, Ser. No. 116,957 proximately 100% sulfuric acid ranges from about 48 3 (Caims. (CE. 23-39) grams per 100 grams of solution at 0° C. up to about 68 grams at 50 C. with, of course, a lesser solubility below This invention relates to a proces for the production of O 0 C. and a greater above 50° C. Although the presence nitrosylsulfuric acid. of the precipitated nitrosylsulfuric acid is usually a source Nitrosyisulfuric acid is particularly desirable for use of mechanical inconvenience, advantage may be taken of in the production of caprolactam from hexahydrobenzoic it by removal of the solid nitrosylsulfuric acid by filtra acid. Nitrosylsulfuric acid has long been known as an 5 tion and subsequent recycle of the mother liquor to the intermediate in connection with the lead-chamber sulfuric reaction Zone. acid process in which it is converted to Sulfuric acid with in general, the effective temperature range of the process concurrent liberation of nitric oxide in a reaction with is defined by the requirement that the reaction medium be sulfur dioxide. -

New Synthesis Routes for Production of Ε-Caprolactam by Beckmann
New synthesis routes for production of ε-caprolactam by Beckmann rearrangement of cyclohexanone oxime and ammoximation of cyclohexanone over different metal incorporated molecular sieves and oxide catalysts Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTH Aachen University zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigte Dissertation vorgelegt von Anilkumar Mettu aus Guntur/Indien Berichter: Universitätprofessor Dr. Wolfgang F. Hölderich Universitätprofessor Dr. Carsten Bolm Tag der mündlichen Prüfung: 29.01.2009 Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar. Dedicated to my Parents This work reported here has been carried out at the Institute for Chemical Technolgy and Heterogeneous Catalysis der Fakultät für Mathematik, Informatik und Naturwissenschaften in the University of Technology, RWTH Aachen under supervision of Prof. Dr. Wolfgang F. Hölderich between June 2005 and August 2008. ACKNOWLEDGEMENTS I would like to express my deepest sence of gratitude to my supervisor Prof. Dr. rer. nat. W. F. Hölderich for giving me the opportunity to do my doctoral study in his group. His guidance and teaching classes have allowed me to grow and learn my subject during my Ph.d. He has provided many opportunities for me to increase my abilities as a researcher and responsibilities as a team member. I am grateful for the financial support of this work from Sumitomo Chemicals Co., Ltd, Niihama, Japan (Part One) and Uhde Inventa-Fischer GmBH, Berlin (Part Two). Our collaborators at Sumitomo Chemicals Co., Ltd (Dr. C. Stoecker) and Uhde Inventa- Fischer GmBH (Dr. R. Schaller and Dr. A. Pawelski) provided thoughtful guidance and suggestions for each project. -

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11
This PDF is available from The National Academies Press at http://www.nap.edu/catalog.php?record_id=13374 Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11 ISBN Committee on Acute Exposure Guideline Levels; Committee on 978-0-309-25481-6 Toxicology; National Research Council 356 pages 6 x 9 PAPERBACK (2012) Visit the National Academies Press online and register for... Instant access to free PDF downloads of titles from the NATIONAL ACADEMY OF SCIENCES NATIONAL ACADEMY OF ENGINEERING INSTITUTE OF MEDICINE NATIONAL RESEARCH COUNCIL 10% off print titles Custom notification of new releases in your field of interest Special offers and discounts Distribution, posting, or copying of this PDF is strictly prohibited without written permission of the National Academies Press. Unless otherwise indicated, all materials in this PDF are copyrighted by the National Academy of Sciences. Request reprint permission for this book Copyright © National Academy of Sciences. All rights reserved. Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11 Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies Copyright © National Academy of Sciences. All rights reserved. Acute Exposure Guideline Levels for Selected Airborne Chemicals: Volume 11 THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Insti- tute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. -

AP Chemistry Free Response
AP Chemistry Exam Reactions: Questions and Answers With the new format of the exam in 2007 and the availability of both questions and answers on the web at AP Central (http://apcentral.collegeboard.com:80/apc/public/courses/4606.html), I have determined not to update this page any longer. Please create an account as a teacher at AP Central and navigate to the full exams and scoring rubrics which are available back to 2003 Beginning in 2007, question 4 is no longer 5 out of 8 responses but rather three required responses. Also, in addition to writing the reactants and products, the equation must be balanced and there is a question about the chemical reaction. 2007 (a) A solution of sodium hydroxide is added to a solution of lead(II) nitrate. If 1.0 L volumes of 1.0 M solutions of sodium hydroxide and lead(II) nitrate are mixed together, now many moles of product(s) will be produced? Assume the reaction goes to completion. (b) Excess nitric acid is added to solid calcium carbonate. Briefly explain why statues made of marble (calcium carbonate) displayed outdoors in urban areas are deteriorating. (c) A solution containing silver(I) ion (an oxidixing agent) is mixed with a; solution containing iron(II) ion (a reducing agent). If the contents of the reaction mixture described above are filtered, what substance(s), if any, would remain on the filter paper? - 2+ → (a) (i) Balanced equation: 2OH + Pb Pb(OH)2 (s) (ii) The moles of each reactant are obtained by multiplying the volume times the molarity. -

Download Safety Data Sheet (SDS)
SAFETY DATA SHEET 1. Identification Product identifier Strike 80 Soil Fumigant Other means of identification SDS number 180-AUS-TCA Recommended use Soil fumigant NOTE TO PESTICIDE HANDLERS: If the pesticide product end-use labeling contains hazard information, specific instructions, or requirements that conflict with this Safety Data Sheet (SDS), follow the hazard information, instructions, or requirements on the labeling. Restrictions on use Use of this product requires supervision by a qualified pesticide applicator. Details of manufacturer or importer TriCal Australia Pty Ltd Address 5 Chamberlain Street, Wingfield, SA 5013, Australia Telephone 08 8347 3838 E-mail [email protected] Emergency phone number CHEMTREC (Australia) 02 9037 2994 (24/7) POISONS INFORMATION CENTRE 13 11 26 2. Hazard(s) identification Physical hazards Not classified. Health hazards Acute toxicity, oral Category 2 Acute toxicity, dermal Category 2 Acute toxicity, inhalation Category 1 Skin corrosion/irritation Category 1C Serious eye damage/eye irritation Category 1 Sensitization, skin Category 1 Carcinogenicity Category 2 Specific target organ toxicity, Category 1 (respiratory system damage) single exposure Specific target organ toxicity, Category 3 (respiratory tract irritation) single exposure Specific target organ toxicity, Category 1 (respiratory tract/lungs) repeated exposure Environmental hazards Hazardous to the aquatic environment, Category 1 acute hazard Hazardous to the aquatic environment, Category 2 long-term hazard Label elements Skull Corrosion Health Exclamation Environment Signal word DANGER Hazard statement Fatal if swallowed, in contact with skin or if inhaled. May cause an allergic skin reaction. Causes serious eye damage. Causes severe skin burns and eye damage. Strike 80 Soil Fumigant November 25, 2019 Page 1 of 11 May cause respiratory irritation. -

Aug. 29, 1967 Yoshi KAZU ITO ETAL 3,338,887 PREPARATION of NITROSYL CHLORIDE Filed Dec
Aug. 29, 1967 Yoshi KAZU ITO ETAL 3,338,887 PREPARATION OF NITROSYL CHLORIDE Filed Dec. 26, 1962 INVENTORS YOSHIKAZU ITO FUMO NS-KAWA 2%-42.TAKAO WAMURA ATTORNEY 3,338,887 United States Patent Office Patented Aug. 29, 1967 1. 2 3,338,887 be taken out of the cyclic system in order to maintain PREPARATION OF NITROSYL CHLORIDE its material balance, or, alternatively, the water has to Yoshikazu to, Mizuho-ku, Nagoya, and Fumio Nishi be taken out of the cyclic system by distillation of the kawa and Takao Iwamura, Minami-ku, Nagoya, Japan, spent liquor under reduced pressure. assignors to Toyo Rayon Kabushiki Kaisha, Tokyo, Japan, a corporation of Japan While the spent liquor which has been taken out to Filed Dec. 26, 1962, Ser. No. 246,914 the outside of the system by the above method can be Claims priority, application Japan, Dec. 26, 1961, used for other purposes as sulfuric acid by being con 36/46,743 verted thereto by the conventional nitric oxide process 1 Claim. (Cl. 260-239.3) for sulfuric acid manufacture, the recovery of the oxides 10 of nitrogen which are formed in this instance is a dis This invention relates to a method of preparing nitro advantage from the commercial standpoint. Furthermore, syl chloride which makes it possible to prepare nitrosyl since a small amount of hydrochloric acid is contained chloride cyclically with advantage and effectiveness on in the cycling spent liquor, hydrochloric acid is also a commercial scale, and in which the liquid portion which contained in the sulfuric acid formed. -
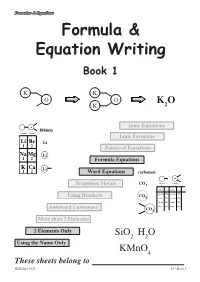
Formula & Equation Writing
Formulae & Equations Formula & Equation Writing Book 1 K K O O K O K 2 Ionic Equations lithium column column 1 2 Ionic Formulae Li Be Li 1 2 Balanced Equations Na Mg Li 1 2 Formula Equations K Ca Li 1 2 Word Equations carbonate valency valency Transition Metals CO3 1 2 name formula name formula ammonium NH4 carbonate CO3 Using Brackets CO 3 cyanide CN chromate CrO4 hydroxide OH sulphate SO4 nitrate NO sulphite SO Awkward Customers 3 3 CO3 More than 2 Elements 2 Elements Only SiO2 H2O Using the Name Only KMnO4 These sheets belong to KHS Sept 2013 page 1 S3 - Book 1 Formulae & Equations What is a Formula ? The formula of a compound tells you two things about the compound :- SiO H O i) which elements are in the compound 2 2 using symbols, and KMnO4 ii) how many atoms of each element are in the compound using numbers. Test Yourself What would be the formula for each of the following? Br H Cl K H C S Al Br O H Cl K H Br Using the Name Only Some compounds have extra information in their carbon monoxide names that allow people to work out and write CO the correct formula. carbon dioxide CO2 The names of the elements appear as usual but dinitrogen tetroxide N O this time the number of each type of atom is 2 4 included using mono- = 1 di- = 12 tri- = 3 tetra- = 4 penta- = 5 hexa- =46 Test Yourself 1 What would be the formula for each of the following? 1. -

Nitric Acid Plants (PDF)
Office of Air and Radiation December 2010 AVAILABLE AND EMERGING TECHNOLOGIES FOR REDUCING GREENHOUSE GAS EMISSIONS FROM THE NITRIC ACID PRODUCTION INDUSTRY Available and Emerging Technologies for Reducing Greenhouse Gas Emissions from the Nitric Acid Production Industry Prepared by the Sector Policies and Programs Division Office of Air Quality Planning and Standards U.S. Environmental Protection Agency Research Triangle Park, North Carolina 27711 December 2010 Table of Contents Abbreviations and Acronyms p.1 I Introduction /Purpose of this Document p.2 II. Description of the Nitric Acid Production Process p.3 A. Weak Nitric Acid Production p.3 B. High-Strength Nitric Acid Production p.6 III. N2O Emissions and Nitric Acid Production Process p. 7 IV. Summary of Control Measures p.9 A. Primary Controls p.10 B. Secondary Control p.11 C. Tertiary Controls p.13 D. Selective Catalytic Reduction p.16 V. Other Greenhouse Gas Emissions p.16 VI. Energy Efficiency Improvements p.17 VII. EPA Contacts p.19 VIII. References p.20 Appendix A – Southeast Idaho Energy p.23 Appendix B - US Nitric Acid Plants p.24 Appendix C – CDM Monitoring Reports p.25 Abbreviations and Acronyms atm Pressure in atmospheres BACT Best Available Control Technologies Btu British Thermal Unit Btu/lb Btu per lb of 100% nitric acid CAR Climate Action Reserve CDM Clean Development Mechanism CHP Combined Heat and Power CO2 Carbon dioxide CO2e Carbon dioxide equivalent EU European Union GHG Greenhouse Gases H2 Hydrogen IPCC Intergovernmental Panel on Climate Change IPPC Industrial Pollution Prevention and Control JI Joint Implementation kg N2O/tonne kilograms of N2O per tonne of 100% nitric acid kg CO2e/tonne kilograms of CO2e per tonne of 100% nitric acid lb N2O/ton pounds of N2O per ton of 100% nitric acid N2 Nitrogen NH3 Ammonia NO Nitric oxide NO2 Nitrogen dioxide NOx Nitrogen oxides NSCR Nonselective Catalytic Reduction N2 O4 Nitrogen tetraoxide N2O Nitrous oxide O2 Oxygen PSD Prevention of Significant Deterioration SCR Selective Catalytic Reduction TPD Tons per day 1 I. -

COVALENT COMPOUNDS Unit 2 COVALENT BOND
COVALENT COMPOUNDS Unit 2 COVALENT BOND Forms between two non-metals! Electrons are shared! Co= share Valent = electrons Since electrons are shared, no need to balance charges Covalent bonds form molecules. Molecule - group of neutral atoms held together with a covalent bond Diatomic molecules – molecule consisting of 2 atoms MOLECULES H2O2Br2F2I2N2Cl2 Compound composed of molecules is a molecular compound. MOLECULAR COMPOUNDS Molecular Compounds Properties Gases or liquids Low melting/boiling points Usually made from 2 nonmetals bonded covalently Do not conduct electricity (solid or dissolved) Molecular chemical formula shows how many atoms of each element a molecule contains. H2O – Molecular formula of water 2 Hydrogen atoms, 1 Oxygen atom CO2 – Molecular formula for Carbon Dioxide MOLECULAR 1 Carbon atom, 2 Oxygen atoms FORMULAS H:H - Lewis Dot H-H – Structural Formula H2 – Molecular chemical formula LEWIS STRUCTURES COVALENT BONDING To draw a Lewis Structure, one must know the types of atoms in the molecule, the number of atoms in the molecule and the number of valence electrons for each atom in the molecule. DOUBLE & TRIPLE COVALENT BONDS Sharing a pair of electrons creates a single covalent bond Sharing more than one pair of electrons creates double and triple covalent bonds. 2 shared pairs – Double covalent bond 3 shared pairs – Triple covalent bond Carbon, Nitrogen, Oxygen 1. First element is named first, using the full name of the element 2. The second element is named by dropping ending and adding RULES FOR “–ide” NAMING 3. Prefixes are used to denote # COVALENT of each atom present COMPOUNDS 4. Prefix mono- is never used on the first element 5. -
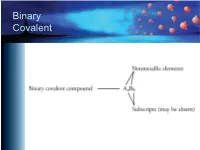
Binary Covalent Common Names
Binary Covalent Common Names – H2O, water – NH3, ammonia – CH4, methane – C2H6, ethane – C3H8, propane – C4H10, butane – C5H12, pentane – C6H14, hexane Naming Binary Covalent CompounDs • If the subscript for the first element is greater than one, inDicate the subscript with a prefix. – We Do not write mono- on the first name. – Leave the "a" off the enD of the prefixes that enD in "a" anD the “o” off of mono- if they are placeD in front of an element that begins with a vowel (oxygen or ioDine). Prefixes mon(o) hex(a) di hept(a) tri oct(a) tetr(a) non(a) pent(a) dec(a) Nitrogen OxiDe Names • N2O3 – name starts with di • N2O5 – name starts with di • NO2 – no initial prefix • NO – no initial prefix Naming Binary Covalent CompounDs • Follow the prefix with the name of the first element in the formula. – N2O3 – dinitrogen – N2O3 – dinitrogen – NO2 – nitrogen – NO – nitrogen Naming Binary Covalent CompounDs • Write a prefix to inDicate the subscript for the seconD element. (Remember to leave the “o” off of mono- anD the “a” off of the prefixes that enD in “a” when they are placeD in front of a name that begins with a vowel.) – N2O3 – dinitrogen tri – N2O5 – dinitrogen pent – NO2 – nitrogen di – NO – nitrogen mon Naming Binary Covalent CompounDs • Write the root of the name of the seconD symbol in the formula. (See the next sliDe.) – N2O3 – dinitrogen triox – N2O5 – dinitrogen pentox – NO2 – nitrogen diox – NO – nitrogen monox Roots of Nonmetals H hydr- F fluor- C carb- Cl chlor- N nitr- Br brom- P phosph- I iod- O ox- S sulf- Se selen- Naming Binary Covalent CompounDs • AdD -ide to the enD of the name. -

Modeling the Vapor – Liquid Equilibrium and Association
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Open Archive Toulouse Archive Ouverte MODELING THE VAPOR – LIQUID EQUILIBRIUM AND ASSOCIATION OF NITROGEN DIOXIDE / DINITROGEN TETROXIDE AND ITS MIXTURES WITH CARBON DIOXIDE A. Belkadi 1,2 , F. Llovell 3, V. Gerbaud 1*, L. F. Vega 2,3*,+ 1Université de Toulouse, LGC (Laboratoire de Génie Chimique), CNRS, INP, UPS, 5 rue Paulin Talabot, F-31106 Toulouse Cedex 01 – France 2MATGAS Research Center, Campus UAB. 08193 Bellaterra, Barcelona Spain 3Institut de Ciència de Materials de Barcelona, (ICMAB-CSIC), Consejo Superior de Investigaciones Científicas, Campus de la UAB, 08193 Bellaterra, Barcelona, Spain * Corresponding authors: [email protected], [email protected]. +Present address: Carburos Metálicos-Grup Air Products. C/ Aragón 300. 08009 Barcelona. Spain. 1 Abstract We have used in this work the crossover soft-SAFT equation of state to model nitrogen dioxide/dinitrogen tetraoxide (NO 2/N 2O4), carbon dioxide (CO 2) and their mixtures. The prediction of the vapor – liquid equilibrium of this mixture is of utmost importance to correctly assess the NO 2 monomer amount that is the oxidizing agent of vegetal macromolecules in the CO 2 + NO 2 / N 2O4 reacting medium under supercritical conditions. The quadrupolar effect was explicitly considered when modeling carbon dioxide, enabling to obtain an excellent description of the vapor-liquid equilibria diagrams. NO 2 was modeled as a self associating molecule with a single association site to account for the strong associating character of the NO 2 molecule. Again, the vapor- liquid equilibrium of NO 2 was correctly modeled.