Clustering of the Structures of Protein Kinase Activation Loops
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
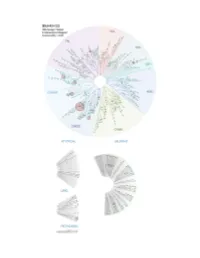
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

Silencing of Lncrna H19 Enhances the Sensitivity to X-Rays and Carbon-Ions Through the Mir-130A-3P /WNK3 Signal Axis in Non-Small-Cell Lung Cancer Cells
Silencing of lncRNA H19 Enhances the Sensitivity to X-rays and Carbon-Ions Through the miR-130a-3p /WNK3 Signal Axis in Non-Small-Cell Lung Cancer Cells Xueshan Zhao Lanzhou University First Aliated Hospital https://orcid.org/0000-0002-1194-7617 Xiaodong Jin Institute of Modern Physics Chinese Academy of Sciences Qiuning Zhang Institute of Modern Physics Chinese Academy of Sciences Ruifeng Liu Institute of Modern Physics Chinese Academy of Sciences Hongtao Luo Institute of Modern Physics Chinese Academy of Sciences Zhen Yang Lanzhou University School of Basic Medical Sciences Yichao Geng Lanzhou University First Aliated Hospital Shuangwu Feng Lanzhou University First Aliated Hospital Chengcheng Li Lanzhou University First Aliated Hospital Lina Wang Lanzhou University First Aliated Hospital Xiaohu Wang ( [email protected] ) Lanzhou University First Aliated Hospital Qiang Li Institute of Modern Physics Chinese Academy of Sciences Research Article Keywords: LncRNA H19, MiR-130a-3p, WNK3, Non-small-cell lung cancer, Radiotherapy Posted Date: August 10th, 2021 Page 1/20 DOI: https://doi.org/10.21203/rs.3.rs-768334/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/20 Abstract Background: LncRNA H19 was believed to act as an oncogene in various types of tumors and was considered to be a therapeutic target and diagnosis marker. However, the role of lncRNA H19 in regulating the radiosensitivity of non-small cell lung cancer (NSCLC) cells was unknown. However, the effects of lncRNA H19 on radiosensitivity of NSCLC were not clear. Methods: The expression proles of lncRNAs were explored via transcriptome sequencing in NSCLC. -

A Minor Role of WNK3 in Regulating Phosphorylation of Renal NKCC2 and NCC Co-Transporters in Vivo
120 Research Article A minor role of WNK3 in regulating phosphorylation of renal NKCC2 and NCC co-transporters in vivo Katsuyuki Oi1, Eisei Sohara1,*, Tatemitsu Rai1, Moko Misawa1, Motoko Chiga1, Dario R. Alessi2, Sei Sasaki1 and Shinichi Uchida1 1Department of Nephrology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8519, Japan 2MRC Protein Phosphorylation Unit, College of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, Scotland, UK *Author for correspondence ([email protected]) Biology Open 1, 120–127 doi: 10.1242/bio.2011048 Summary Mutations in WNK1 and WNK4 kinase genes have been knockout mice. Na+ and K+ excretion in urine in WNK3 shown to cause a human hereditary hypertensive disease, knockout mice was not affected under different salt diets. pseudohypoaldosteronism type II (PHAII). We previously Blood pressure in WNK3 knockout mice was not lower under discovered that WNK kinases phosphorylate and activate normal diet. However, lower blood pressure was observed in OSR1/SPAK kinases that regulate renal SLC12A family WNK3 knockout mice fed low-salt diet. WNK4 and WNK1 transporters such as NKCC2 and NCC, and clarified that the expression was slightly elevated in the knockout mice under constitutive activation of this cascade causes PHAII. WNK3, low-salt diet, suggesting compensation for WNK3 knockout another member of the WNK kinase family, was reported to by these WNKs. Thus, WNK3 may have some role in the be a strong activator of NCC/NKCC2 when assayed in WNK-OSR1/SPAK-NCC/NKCC2 signal cascade in the Xenopus oocytes, suggesting that WNK3 also plays a major kidney, but its contribution to total WNK kinase activity role in regulating blood pressure and sodium reabsorption in may be minimal. -

Comparative Transcriptome Profiling of Selected Osmotic Regulatory Proteins in the Gill During Seawater Acclimation of Chum Salm
www.nature.com/scientificreports OPEN Comparative transcriptome profling of selected osmotic regulatory proteins in the gill during seawater acclimation of chum salmon (Oncorhynchus keta) fry Sang Yoon Lee 1, Hwa Jin Lee2 & Yi Kyung Kim1,2* Salmonid fshes, chum salmon (Oncorhynchus keta) have the developed adaptive strategy to withstand wide salinity changes from the early life stage. This study investigated gene expression patterns of cell membrane proteins in the gill of chum salmon fry on the transcriptome level by tracking the salinity acclimation of the fsh in changing environments ranging from freshwater (0 ppt) to brackish water (17.5 ppt) to seawater (35 ppt). Using GO analysis of DEGs, the known osmoregulatory genes and their functional groups such as ion transport, transmembrane transporter activity and metal ion binding were identifed. The expression patterns of membrane protein genes, including pump-mediated protein (NKA, CFTR), carrier-mediated protein (NKCC, NHE3) and channel-mediated protein (AQP) were similar to those of other salmonid fshes in the smolt or adult stages. Based on the protein-protein interaction analysis between transmembrane proteins and other related genes, we identifed osmotic-related genes expressed with salinity changes and analyzed their expression patterns. The fndings of this study may facilitate the disentangling of the genetic basis of chum salmon and better able an understanding of the osmophysiology of the species. Salinity is one of the critical factors limiting the distribution patterns of all aquatic organisms1–4. Salmonid fshes display diverse life-history traits; anadromous individuals that mature in the river from hatching through to juveniles acquire the capacity to tolerate salinity associated with parr–smolt transformation and undergo ocean migrations before returning to rivers for spawning, whereas landlocked types spend their entire life within fresh- water5,6. -

Common Noncoding UMOD Gene Variants Induce Salt
LETTERS Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression Matteo Trudu1, Sylvie Janas2,3, Chiara Lanzani4, Huguette Debaix2,3, Céline Schaeffer1, Masami Ikehata5,6, Lorena Citterio4, Sylvie Demaretz7, Francesco Trevisani8, Giuseppe Ristagno9, Bob Glaudemans2, Kamel Laghmani7, Giacomo Dell’Antonio10, the Swiss Kidney Project on Genes in Hypertension (SKIPOGH) team11, Johannes Loffing12, Maria P Rastaldi5,6, Paolo Manunta8, Olivier Devuyst2,3,13 & Luca Rampoldi1,13 Hypertension and chronic kidney disease (CKD) are complex population-based association studies12–14. However, deciphering the traits representing major global health problems1,2. Multiple biological mechanisms underlying these genetic associations has genome-wide association studies have identified common proven to be a major challenge. variants in the promoter of the UMOD gene3–9, which encodes Recent genome-wide association studies (GWAS) in more than uromodulin, the major protein secreted in normal urine, that 200,000 individuals of European ancestry have identified susceptibil- cause independent susceptibility to CKD and hypertension. ity variants for renal function, CKD and hypertension in the UMOD Despite compelling genetic evidence for the association gene encoding uromodulin3–9. Uromodulin (or Tamm-Horsfall pro- between UMOD risk variants and disease susceptibility in tein) is the most abundant urinary protein and is specifically pro- the general population, the underlying biological mechanism duced and secreted by the epithelial cells lining the thick ascending is not understood. Here, we demonstrate that UMOD risk limb (TAL) of the loop of Henle in the kidney15. Studies in Umod variants increased UMOD expression in vitro and in vivo. knockout mice revealed that uromodulin may protect against urinary Uromodulin overexpression in transgenic mice led to salt- tract infection16 and kidney stones17 and modulate electrolyte tubu- sensitive hypertension and to the presence of age-dependent lar transport18. -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes
JASN Express. Published on December 3, 2008 as doi: 10.1681/ASN.2008040406 BASIC RESEARCH www.jasn.org Large-Scale Proteomics and Phosphoproteomics of Urinary Exosomes Patricia A. Gonzales,*† Trairak Pisitkun,* Jason D. Hoffert,* Dmitry Tchapyjnikov,* ʈ Robert A. Star,‡ Robert Kleta,§ ¶ Nam Sun Wang,† and Mark A. Knepper* *Laboratory of Kidney and Electrolyte Metabolism, National Heart, Lung, and Blood Institute, ‡Renal Diagnostics and Therapeutics Unit, National Institute of Diabetes and Digestive and Kidney Diseases, §Section of Human ʈ Biochemical Genetics, Medical Genetics Branch, National Human Genome Research Institute, and Office of Rare Diseases, Office of the Director, National Institutes of Health, Bethesda, and †Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, Maryland; and ¶London Epithelial Group, Centre for Nephrology, University College London, London, United Kingdom ABSTRACT Normal human urine contains large numbers of exosomes, which are 40- to 100-nm vesicles that originate as the internal vesicles in multivesicular bodies from every renal epithelial cell type facing the urinary space. Here, we used LC-MS/MS to profile the proteome of human urinary exosomes. Overall, the analysis identified 1132 proteins unambiguously, including 177 that are represented on the Online Mendelian Inheritance in Man database of disease-related genes, suggesting that exosome analysis is a potential approach to discover urinary biomarkers. We extended the proteomic analysis to phospho- proteomic profiling using neutral loss scanning, and this yielded multiple novel phosphorylation sites, including serine-811 in the thiazide-sensitive Na-Cl co-transporter, NCC. To demonstrate the potential use of exosome analysis to identify a genetic renal disease, we carried out immunoblotting of exosomes from urine samples of patients with a clinical diagnosis of Bartter syndrome type I, showing an absence of the sodium-potassium-chloride co-transporter 2, NKCC2. -
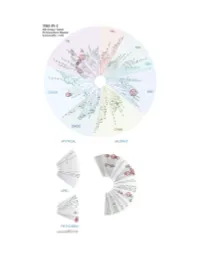
Profiling Data
Entrez Gene Percent Compound Compound Name DiscoveRx Gene Symbol Symbol Control Concentration (nM) THZ-P1-2 AAK1 AAK1 100 1000 THZ-P1-2 ABL1(E255K)-phosphorylated ABL1 21 1000 THZ-P1-2 ABL1(F317I)-nonphosphorylated ABL1 93 1000 THZ-P1-2 ABL1(F317I)-phosphorylated ABL1 100 1000 THZ-P1-2 ABL1(F317L)-nonphosphorylated ABL1 71 1000 THZ-P1-2 ABL1(F317L)-phosphorylated ABL1 44 1000 THZ-P1-2 ABL1(H396P)-nonphosphorylated ABL1 5.8 1000 THZ-P1-2 ABL1(H396P)-phosphorylated ABL1 6.6 1000 THZ-P1-2 ABL1(M351T)-phosphorylated ABL1 12 1000 THZ-P1-2 ABL1(Q252H)-nonphosphorylated ABL1 24 1000 THZ-P1-2 ABL1(Q252H)-phosphorylated ABL1 20 1000 THZ-P1-2 ABL1(T315I)-nonphosphorylated ABL1 93 1000 THZ-P1-2 ABL1(T315I)-phosphorylated ABL1 100 1000 THZ-P1-2 ABL1(Y253F)-phosphorylated ABL1 2.4 1000 THZ-P1-2 ABL1-nonphosphorylated ABL1 13 1000 THZ-P1-2 ABL1-phosphorylated ABL1 8.1 1000 THZ-P1-2 ABL2 ABL2 36 1000 THZ-P1-2 ACVR1 ACVR1 94 1000 THZ-P1-2 ACVR1B ACVR1B 100 1000 THZ-P1-2 ACVR2A ACVR2A 94 1000 THZ-P1-2 ACVR2B ACVR2B 91 1000 THZ-P1-2 ACVRL1 ACVRL1 90 1000 THZ-P1-2 ADCK3 CABC1 77 1000 THZ-P1-2 ADCK4 ADCK4 97 1000 THZ-P1-2 AKT1 AKT1 95 1000 THZ-P1-2 AKT2 AKT2 95 1000 THZ-P1-2 AKT3 AKT3 100 1000 THZ-P1-2 ALK ALK 92 1000 THZ-P1-2 ALK(C1156Y) ALK 93 1000 THZ-P1-2 ALK(L1196M) ALK 71 1000 THZ-P1-2 AMPK-alpha1 PRKAA1 93 1000 THZ-P1-2 AMPK-alpha2 PRKAA2 100 1000 THZ-P1-2 ANKK1 ANKK1 97 1000 THZ-P1-2 ARK5 NUAK1 83 1000 THZ-P1-2 ASK1 MAP3K5 100 1000 THZ-P1-2 ASK2 MAP3K6 95 1000 THZ-P1-2 AURKA AURKA 99 1000 THZ-P1-2 AURKB AURKB 100 1000 THZ-P1-2 AURKC AURKC 83 1000 -

Gene Symbol Accession Alias/Prev Symbol Official Full Name AAK1 NM 014911.2 KIAA1048, Dkfzp686k16132 AP2 Associated Kinase 1
Gene Symbol Accession Alias/Prev Symbol Official Full Name AAK1 NM_014911.2 KIAA1048, DKFZp686K16132 AP2 associated kinase 1 (AAK1) AATK NM_001080395.2 AATYK, AATYK1, KIAA0641, LMR1, LMTK1, p35BP apoptosis-associated tyrosine kinase (AATK) ABL1 NM_007313.2 ABL, JTK7, c-ABL, p150 v-abl Abelson murine leukemia viral oncogene homolog 1 (ABL1) ABL2 NM_007314.3 ABLL, ARG v-abl Abelson murine leukemia viral oncogene homolog 2 (arg, Abelson-related gene) (ABL2) ACVR1 NM_001105.2 ACVRLK2, SKR1, ALK2, ACVR1A activin A receptor ACVR1B NM_004302.3 ACVRLK4, ALK4, SKR2, ActRIB activin A receptor, type IB (ACVR1B) ACVR1C NM_145259.2 ACVRLK7, ALK7 activin A receptor, type IC (ACVR1C) ACVR2A NM_001616.3 ACVR2, ACTRII activin A receptor ACVR2B NM_001106.2 ActR-IIB activin A receptor ACVRL1 NM_000020.1 ACVRLK1, ORW2, HHT2, ALK1, HHT activin A receptor type II-like 1 (ACVRL1) ADCK1 NM_020421.2 FLJ39600 aarF domain containing kinase 1 (ADCK1) ADCK2 NM_052853.3 MGC20727 aarF domain containing kinase 2 (ADCK2) ADCK3 NM_020247.3 CABC1, COQ8, SCAR9 chaperone, ABC1 activity of bc1 complex like (S. pombe) (CABC1) ADCK4 NM_024876.3 aarF domain containing kinase 4 (ADCK4) ADCK5 NM_174922.3 FLJ35454 aarF domain containing kinase 5 (ADCK5) ADRBK1 NM_001619.2 GRK2, BARK1 adrenergic, beta, receptor kinase 1 (ADRBK1) ADRBK2 NM_005160.2 GRK3, BARK2 adrenergic, beta, receptor kinase 2 (ADRBK2) AKT1 NM_001014431.1 RAC, PKB, PRKBA, AKT v-akt murine thymoma viral oncogene homolog 1 (AKT1) AKT2 NM_001626.2 v-akt murine thymoma viral oncogene homolog 2 (AKT2) AKT3 NM_181690.1 -

Anatomy of a Pressure Sensing Protein Kinase Authors
bioRxiv preprint doi: https://doi.org/10.1101/435008; this version posted October 4, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Anatomy of a pressure sensing protein kinase Authors: Radha Akella1, Kamil Sekulski1, John M. Pleinis2, Joanna Liwocha1, Jenny Jiou1, Haixia He1, John M. Humphreys1, Jeffrey N. Schellinger3, Lucasz Joachimiak4, Melanie Cobb5, Aylin R. Rodan2, and Elizabeth J. Goldsmith1∗ Affiliations: 1 Department of Biophysics, The University of Texas Southwestern Medical Center at Dallas, 5323 Harry Hines Boulevard,Dallas, Texas 75390-8816. 2Department of Internal Medicine, Division of Nephrology and Hypertension and Molecular Medicine Program, The University of Utah, 15North 2030 East Salt Lake City Utah 84112 . 3Division of Nephrology, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas.75390. 4Center for Alzheimer’s and Neurodegenerative Diseases, Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, Texas.75390 5Department of Pharmacology, University of Texas Southwestern Medical Center, Dallas, Texas.75390 *Correspondence to: [email protected] Abstract Cells respond to hydrostatic pressure to maintain cellular, organ and organism level functions, to set and respond to blood pressure, tissue perfusion, filtration rates and other processes. Pressure sensing is thought to occur at membranes where proteins can respond to mechanical cues1,2,3 . Thus, proteins implicated as direct pressure sensors have been mainly ion channels 2,4, and more recently G-protein coupled receptors5,6. Here we show, contrary to expectations, that hydrostatic pressure directly induces autophosphorylation and activation of an intracellular protein kinase, With No Lysine kinase-3 (WNK3), and to a lesser extent, WNK17. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein