Synopsis GRC 4039-204
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sadhanayatra
SADHANA YATRA December 23, 2012~ January 1o, 2013 {option to start on December 27, 2012} {optional extension January 11 - 12, 2013} JOIN NĀMARŪPA & FRIENDS ON A PILGRIMAGE TO INDIA Chennai Beach Resort ~ Tiruvannamalai Retreat with Swamijis from Himalayas Chidambaram Festival ~ Puducherry ~ Kanchipuram ~ Tirupati ~ Govardhan Eco ~ Village Retreat ~ Ashta Vinayaka ~ Pandharpur ~ Trimbakeshwar ~ Mumbai Sunday December 23, 2012 ~ Chennai MGM Beach Resort Prithivi (earth) lingam and Varadharaja Perumal (Lord Vishnu). Arrive in Chennai no later than the night of December 22 or early Tuesday January 1, 2013 ~ Tirupati morning on December 23. Check into MGM Beach Resort. Relax. Depart for Sri Kalahasthi and darshan of Vayu (air) Lingam, and then Meet the group & discuss the Yatra, daily programs, & local customs. on to Tirupati. Monday December 24 ~ Tiruvannamalai Retreat Wednesday January 2 ~ Tirupati After a late morning's rest we will travel through the beautiful South Climb 4000 steps in 4 hours (optional travel by bus) to have darshan Indian rice paddies and ancient boulder formations to the temple of Lord Venkateswara (Balaji). We will take the bus back down! town of Tiruvannamalai and check into the Sparsha Eco-Resort. Thursday January 3 ~ Fly to Mumbai ~ Govardhan Eco Village Tuesday December 25 ~ Tiruvannamalai Retreat A short flight north to Mumbai and then on to the Govardhan Eco During our stay here we will gather together for informal discussions Village for a Bhakti Retreat. with Swamijis from the Himalayas. The swamis are specially gather- Friday January 4 ~ Govardhan Eco Village ing here to greet and talk with us. We will also go for darshan of the Take a well earned rest in the quiet natural setting of the Sahyadri Agni (fire) lingam in the great temple of Arunachaleswara and climb mountains. -

3.Hindu Websites Sorted Country Wise
Hindu Websites sorted Country wise Sl. Reference Country Broad catergory Website Address Description No. 1 Afghanistan Dynasty http://en.wikipedia.org/wiki/Hindushahi Hindu Shahi Dynasty Afghanistan, Pakistan 2 Afghanistan Dynasty http://en.wikipedia.org/wiki/Jayapala King Jayapala -Hindu Shahi Dynasty Afghanistan, Pakistan 3 Afghanistan Dynasty http://www.afghanhindu.com/history.asp The Hindu Shahi Dynasty (870 C.E. - 1015 C.E.) 4 Afghanistan History http://hindutemples- Hindu Roots of Afghanistan whthappendtothem.blogspot.com/ (Gandhar pradesh) 5 Afghanistan History http://www.hindunet.org/hindu_history/mode Hindu Kush rn/hindu_kush.html 6 Afghanistan Information http://afghanhindu.wordpress.com/ Afghan Hindus 7 Afghanistan Information http://afghanhindusandsikhs.yuku.com/ Hindus of Afaganistan 8 Afghanistan Information http://www.afghanhindu.com/vedic.asp Afghanistan and It's Vedic Culture 9 Afghanistan Information http://www.afghanhindu.de.vu/ Hindus of Afaganistan 10 Afghanistan Organisation http://www.afghanhindu.info/ Afghan Hindus 11 Afghanistan Organisation http://www.asamai.com/ Afghan Hindu Asociation 12 Afghanistan Temple http://en.wikipedia.org/wiki/Hindu_Temples_ Hindu Temples of Kabul of_Kabul 13 Afghanistan Temples Database http://www.athithy.com/index.php?module=p Hindu Temples of Afaganistan luspoints&id=851&action=pluspoint&title=H indu%20Temples%20in%20Afghanistan%20. html 14 Argentina Ayurveda http://www.augurhostel.com/ Augur Hostel Yoga & Ayurveda 15 Argentina Festival http://www.indembarg.org.ar/en/ Festival of -
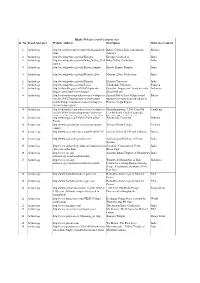
2.Hindu Websites Sorted Category Wise
Hindu Websites sorted Category wise Sl. No. Broad catergory Website Address Description Reference Country 1 Archaelogy http://aryaculture.tripod.com/vedicdharma/id10. India's Cultural Link with Ancient Mexico html America 2 Archaelogy http://en.wikipedia.org/wiki/Harappa Harappa Civilisation India 3 Archaelogy http://en.wikipedia.org/wiki/Indus_Valley_Civil Indus Valley Civilisation India ization 4 Archaelogy http://en.wikipedia.org/wiki/Kiradu_temples Kiradu Barmer Temples India 5 Archaelogy http://en.wikipedia.org/wiki/Mohenjo_Daro Mohenjo_Daro Civilisation India 6 Archaelogy http://en.wikipedia.org/wiki/Nalanda Nalanda University India 7 Archaelogy http://en.wikipedia.org/wiki/Taxila Takshashila University Pakistan 8 Archaelogy http://selians.blogspot.in/2010/01/ganesha- Ganesha, ‘lingga yoni’ found at newly Indonesia lingga-yoni-found-at-newly.html discovered site 9 Archaelogy http://vedicarcheologicaldiscoveries.wordpress.c Ancient Idol of Lord Vishnu found Russia om/2012/05/27/ancient-idol-of-lord-vishnu- during excavation in an old village in found-during-excavation-in-an-old-village-in- Russia’s Volga Region russias-volga-region/ 10 Archaelogy http://vedicarcheologicaldiscoveries.wordpress.c Mahendraparvata, 1,200-Year-Old Cambodia om/2013/06/15/mahendraparvata-1200-year- Lost Medieval City In Cambodia, old-lost-medieval-city-in-cambodia-unearthed- Unearthed By Archaeologists 11 Archaelogy http://wikimapia.org/7359843/Takshashila- Takshashila University Pakistan Taxila 12 Archaelogy http://www.agamahindu.com/vietnam-hindu- Vietnam -
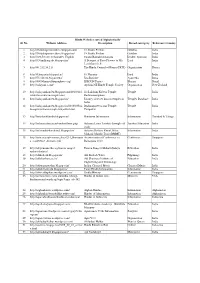
1.Hindu Websites Sorted Alphabetically
Hindu Websites sorted Alphabetically Sl. No. Website Address Description Broad catergory Reference Country 1 http://18shaktipeetasofdevi.blogspot.com/ 18 Shakti Peethas Goddess India 2 http://18shaktipeetasofdevi.blogspot.in/ 18 Shakti Peethas Goddess India 3 http://199.59.148.11/Gurudev_English Swami Ramakrishnanada Leader- Spiritual India 4 http://330milliongods.blogspot.in/ A Bouquet of Rose Flowers to My Lord India Lord Ganesh Ji 5 http://41.212.34.21/ The Hindu Council of Kenya (HCK) Organisation Kenya 6 http://63nayanar.blogspot.in/ 63 Nayanar Lord India 7 http://75.126.84.8/ayurveda/ Jiva Institute Ayurveda India 8 http://8000drumsoftheprophecy.org/ ISKCON Payers Bhajan Brazil 9 http://aalayam.co.nz/ Ayalam NZ Hindu Temple Society Organisation New Zealand 10 http://aalayamkanden.blogspot.com/2010/11/s Sri Lakshmi Kubera Temple, Temple India ri-lakshmi-kubera-temple.html Rathinamangalam 11 http://aalayamkanden.blogspot.in/ Journey of lesser known temples in Temples Database India India 12 http://aalayamkanden.blogspot.in/2010/10/bra Brahmapureeswarar Temple, Temple India hmapureeswarar-temple-tirupattur.html Tirupattur 13 http://accidentalhindu.blogspot.in/ Hinduism Information Information Trinidad & Tobago 14 http://acharya.iitm.ac.in/sanskrit/tutor.php Acharya Learn Sanskrit through self Sanskrit Education India study 15 http://acharyakishorekunal.blogspot.in/ Acharya Kishore Kunal, Bihar Information India Mahavir Mandir Trust (BMMT) 16 http://acm.org.sg/resource_docs/214_Ramayan An international Conference on Conference Singapore -

The Vithoba Temple of Pan<Jharpur and Its Mythological Structure
Japanese Journal of Religious Studies 19S8 15/2-3 The Vithoba Faith of Mahara§tra: The Vithoba Temple of Pan<Jharpur and Its Mythological Structure SHIMA Iwao 島 岩 The Bhakti Movement in Hinduism and the Vithobd Faith o f MaM rdstra1 The Hinduism of India was formed between the sixth century B.C. and the sixth century A.D., when Aryan Brahmanism based on Vedic texts incor porated non-Aryan indigenous elements. Hinduism was established after the seventh century A.D. through its resurgence in response to non-Brah- manistic traditions such as Buddhism. It has continuously incorporated different and new cultural elements while transfiguring itself up to the present day. A powerful factor in this formation and establishment of Hinduism, and a major religious movement characteristic especially of medieval Hin duism, is the bhakti movement (a religious movement in which it is believed that salvation comes through the grace of and faith in the supreme God). This bhakti movement originated in the Bhagavad GUd of the first century B.C., flourishing especially from the middle of the seventh to the middle of the ninth century A.D. among the religious poets of Tamil, such as Alvars, in southern India. This bhakti movement spread throughout India through its incorporation into the tradition of Brahmanism, and later developed in three ways. First, the bhakti movement which had originated in the non-Aryan cul ture of Tamil was incorporated into Brahmanism. Originally bhakti was a strong emotional love for the supreme God, and this was reinterpreted to 1 In connection with this research, I am very grateful to Mr. -

Hindu Dharma
HINDU DHARMA BY M. K. GANDID NAVAJIVAN PUBLISHING HOUSE AHMEDABAD ONAS T IC IB •• A.~¥ Abbey of Ge t h sem ~ I Jrappist, Kentucky HINDU DHARMA M. K. GANDHI NAVAJIVAN PUBLISillNG HOUSE AJIMEDABAD First Edition, 5,000 Copies, August, 1950 J. Reprint, 5,000 Copies, December, 1958 Rupees Four 1 ( ( @ Copyright by the Navajivan Trust, 1950 I I 1 I I ( I I ]A I I ~ " MONASTIC LIBRARY Abbey of Gethseman i Trappist, Kentuck, 'I 'I 'I '1 v Printed and Published by Jivanji Dahyabhai Desai, Navajivan Press, Ahmedabad-14 CONTENTS SECTION ONE : mNDUISM (GENERAL) Chapter Page PUBLISHERS' NOTE iii EDITOR'S NOTE v NON-ENGLISH WORDS WITH THEIR MEANINGS viii 1 GANDHISM 3 2 WHAT IS HINDUISM? 4 3 HINDUISM ALL-INCLUSIVE 4 4 WHY I AM A HINDU 5 5 SANATANA HINDUISM 6 6 SANATANA HINDU 9 7 THE RELIGION OF SERVICE 10 8 HINDUISM NOT A MATTER OF OUTWARD OBSERVANCES 12 9 MY MISSION 13 10 ARYA SAMAJ 14 11 CONVERSION MOVEMENT IN HINDUISM 15 12 WHAT MAY HINDUS DO? 16 13 ORGANIZATION FOR PROTECTING HINDUS 17 14 KRISHNA AND THE MAHABHARATA 18 15 HINDUISM AS EVER GROWING 19 16 THE AUTHORITY OF RELIGIOUS BOOKS 21 17 WHAT IS HINDUISM? 22 18 TULSIDAS 23 19 HINDUISM AND OBSCENITY 25 20 COMMUNICATIONS WITH SPIRITS 26 21 TRUE SHRADDHA 27 22 DIVALI HOLIDAYS 28 23 DUTY OF CEYLON HINDUS 29 24 HELPFUL SPIRITUAL PRACTICES AND READINGS 33 25 EQUALITY IN HINDUISM 35 26 THE CHIEF VALUE OF HINDUISM 35 27 THE GOLDEN KEY 36 28 THE HARIPAD SPEECH 39 29 FROM THE KOTTAYAM SPEECH 41 30 LIVING UP TO 125 43 31 YAJNA OR SACRIFICE H 32 PRAYER DISCOURSES 47 33 WHAT HINDUISM HAS DONE FOR US 48 34 NATIONHOOD THROUGH HINDUISM 50 35 INDIAN CIVILIZATION 50 36 THE LOIN-CLOTH 52 Xl xii HINDU DHARMA A SECTION TWO : GOD 37 GOD 54 38 ADVAITISM AND GOD 55 39 GOD IS 57 40 TRUTH AND GOD 59 A 41 MEANING OF GOD 62 '42 FINDING GOD 63 B C SECTION THREE: TEMPLE·WORSHIP C 43 APPROACH TEMPLES IN FAITH 64 D 44 TEMPLES NECESSARY 65 D 45 IDOL-WORSHIP 65 E 46 TEMPLE-WORSHIP 66 47 GOD AND GODS 66 E 48 "A TEMPLE TO GANDHI]I" 69 49 TREE-WORSHIP 70 F • 50 TEMPLE-WORSHIP 71 G 51 ARE TEMPLES NECESSARY? 73 52 A MODEL TEMPLE 76 f. -
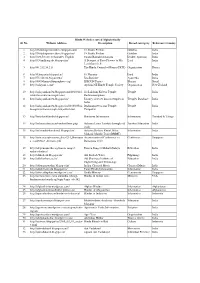
Hindu Websites Sorted Alphabetically Sl
Hindu Websites sorted Alphabetically Sl. No. Website Address Description Broad catergory Reference Country 1 http://18shaktipeetasofdevi.blogspot.com/ 18 Shakti Peethas Goddess India 2 http://18shaktipeetasofdevi.blogspot.in/ 18 Shakti Peethas Goddess India 3 http://199.59.148.11/Gurudev_English Swami Ramakrishnanada Leader- Spiritual India 4 http://330milliongods.blogspot.in/ A Bouquet of Rose Flowers to My Lord India Lord Ganesh Ji 5 http://41.212.34.21/ The Hindu Council of Kenya (HCK) Organisation Kenya 6 http://63nayanar.blogspot.in/ 63 Nayanar Lord India 7 http://75.126.84.8/ayurveda/ Jiva Institute Ayurveda India 8 http://8000drumsoftheprophecy.org/ ISKCON Payers Bhajan Brazil 9 http://aalayam.co.nz/ Ayalam NZ Hindu Temple Society Organisation New Zealand 10 http://aalayamkanden.blogspot.com/2010/11/s Sri Lakshmi Kubera Temple, Temple India ri-lakshmi-kubera-temple.html Rathinamangalam 11 http://aalayamkanden.blogspot.in/ Journey of lesser known temples in Temples Database India India 12 http://aalayamkanden.blogspot.in/2010/10/bra Brahmapureeswarar Temple, Temple India hmapureeswarar-temple-tirupattur.html Tirupattur 13 http://accidentalhindu.blogspot.in/ Hinduism Information Information Trinidad & Tobago 14 http://acharya.iitm.ac.in/sanskrit/tutor.php Acharya Learn Sanskrit through self Sanskrit Education India study 15 http://acharyakishorekunal.blogspot.in/ Acharya Kishore Kunal, Bihar Information India Mahavir Mandir Trust (BMMT) 16 http://acm.org.sg/resource_docs/214_Ramayan An international Conference on Conference Singapore -

CHAPTER 7- Religion and Gods of Maharashtra
Tran DF sfo P rm Y e Y r B 2 B . 0 A Click here to buy w w m w co .A B BYY. CHAPTER 7—RELIGION AND GODS OF MAHARASHTRA IN MAHARASHTRA A MAJORTY OF PEOPLE OF ALL CASTES worship as family deity either one or two of the following Gods : (1) The Mother—goddess. (2) Shiva (3) Khandoba. The fourth God is Vithoba. He is worshipped and revered by most Marathi people but he is not the family deity of many families. Another very popular God is Maruti. The God is known as “ Hanumanta” in Maharashta, Karnatak and Andhra Pradesh, as “ Anjaneya “ in Tamilnad and “ Mahabir “ in the north. He is, however, never a family deity. Other Gods, besides these, are Vishnu, with his incarnations of Ram, Krishna, Narsimha, in rare cases Vamana, A count taken while doing field work in Satara showed that only a minority of people belonging to Brahmin, Maratha, and Mali castes claimed Rama, Krishna, Narsimha as their family deities. Of these again a small number only are worshippers of Narsimha. Narsimha used to be not an uncommon given name in Andhra, Karnatak and Maharashtra1. Dr. S. V. Ketkar wrote that there was an ancient King of Maharashtra called Narsimha perhaps then as later, parts of Maharashtra, Andhra and Karnatak were under one rule. The name would change to Narsu or Narsia in common parlance. It is possible that Narsimha was a legendary hero in this area. He is also connected in a legend of the Chenchu, a tribe from Andhra. People worship many gods Worshippers of Shiva also worship Vishnu. -

Indian Archaeology 1987-88 a Review
INDIAN ARCHAEOLOGY 1987-88 —A REVIEW EDITED BY M.C.JOSHI Director General Archaeological Survey of India PUBLISHED BY THE DIRECTOR GENERAL ARCHAEOLOGICAL SURVEY OF INDIA NEW DELHI 1993 Cover : TisseruStupa,Leh,Ladakh Cover layout & design: Raj Nath Kaw 1992 ARCHAEOLOGICAL SURVEY OF INDIA GOVERNMENT OF INDIA Price :Rs. 250.00 PRINTED AT BENGAL OFFSET WORKS, 335, KHAJOOR ROAD, KAROL BAGH, NEW DELHI-1 10005. PHONE: 524200,7510455 PREFACE I am happy to place before the scholars the Indian Archaeology 1987-88—A Review not very long after the publication of the previous issue. We are making efforts to bring the publication of the Review up to date; this, however, would depend on timely submission of material by contributors. It has been our experience that, in many cases, several reminders have to be given for sending material to us for inclusion in the Review which results in delay of its publication. I am sure, with the cooperation of different institutions and the State Departments of Archaeology and Museums and my own colleagues in different Branches and Circles of the Survey it would be possible for us to maintain regularity of its publication. The present issue of the Review is more voluminous than the previous ones and thus reflects also increased activities in different fields of archaeology. Out of several excavations reported in this issue, I may mention the work at Banawali, Thanesar, Sanghol, Hampi, Sannathi, Daulatabad, Lalitagiri, Udaigiri, Fatehpur Sikri, Sravasti, Balikeshwar, Chandel, Harsud, etc. by the Survey, at Kuntasi jointly by the Deccan College, Pune, and the Gujarat State Department of Archaeology, at Mangalkot by the University of Calcutta, at Kotasur by the Visvabharati, Maihar by the Allahabad University, Ganeshwar by the Department of Archaeology and Museums, Rajasthan State and at Shikarpur by the Department of Archaeology, Government of Gujarat. -

My Experiences of Indian Temples
1 My experiences of Indian temples Anil K. Rajvanshi [email protected] I am not at all a temple-goer and neither do I have an aversion nor particular attraction for temples. Nevertheless as all Indians do, I have also visited many famous temples of India from my childhood till now. I just feel that when millions of people, through ages, have visited them there must be some presence in these temples. There is thus a desire to visit them and feel the presence. However till today I have never felt it in any temple though in one I did get an uneasy feeling. Going to temple is a subjective matter. The readers may or may not agree with my observations. Yet here they are for whatever they are worth. Most of the temples I have visited have been dirty and noisy and in few of them I was harassed by resident pandas. The first harassment I faced was when I visited the famous Krishna temple in Mathura in January 1973. I was going to Vadodara by train and was passing through Mathura where there was a 4-5 hours train halt. I therefore decided to visit the famous Krishna temple in the heart of Mathura. I still remember it was freezing cold in January and I hired a rickshawallah to show me around. The moment I entered the temple, some 10 pandas surrounded me and each one of them wanted to show me the temple. I was warned by the rickshawallah not to take any one of them since I was more interested in seeing the architecture of the temple rather than doing darshan. -

Ts001 Ts305 Ba001 Ba428 Pe001 Pe201 Sa001 Sa229 Mg001
SUB - INSPECTOR OF EXCISE WRITTEN EXAMINATION SEATING ARRANGEMENT Seat No. Seat No. Sr. No. Taluka Schools Name From To 1 Tiswadi Don Bosco High School, Panaji TS001 TS305 2 Bardez G.S. Amonkar High School, Mapusa BA001 BA428 Bicholim 3 Shree Shantadurga High School, Bhailli Peth Bicholim BS001 BS376 Sattari 4 Pernem Shree Bhagwati High School , Pernem PE001 PE201 Ponda 5 A. J. de Almeida High School , Ponda PD001 PD323 Dharbandora 6 Salcete Mahila Nutan English High School, Margao SA001 SA229 7 Mormugao St. Andrew Institute , Mormugao MG001 MG142 Quepem 8 Chandrabhaga Tukoba High School, Curchorem QS001 QS208 Sanguem 9 Canacona St. Theresa High School, Canacona CA001 CA083 CENTRE-WISE FINAL LIST OF QUALIFIED CANDIDATES FOR WRITTEN EXAM FOR THE POST OF SUB-INSPECTOR OF EXCISE TALUKA : Bardez CENTRE : G.S. Amonkar High School, Mapusa Seat No. Name of Candidate Address H. No- 436, Monti Villa, Araddi - Sangolda, Bhd BA001 Narayan Govind Haldankar St. Diogo Church, Guirim, Bardez - Goa. H. No-1668/1, B.B.Borkar Rd, Nr.Post & BA002 Shramik Shrihari Fotto Telephone Colony, Vidhya nagar - Porvorim, Bardez - Goa. BA003 Damodar Ramkrishna Mandrekar H. No- 366, Titowada - Nerul, Bardez - Goa BA004 Maikash Mangaldas Shirodkar H. No- 397/M, D'Mello - Canca, Bardez - Goa. H. No- 194, A-6, Nr.Ganesh Temple, Mapusa, BA005 Milind Madhukar Parab Bardez - Goa. H. No- 1110, Maina - Socorro, Porvorim, Bardez - BA006 Prashant Baburao Halarnkar Goa. H. No- 80/11, Paithona - Salvador De Mundo, BA007 Pravin Vasu Gaonkar Bardez - Goa. H. No- 367, Virlosa, Penha De Franca, Nr BA008 Sagar Vishant Naik Hanuman Temple, Bardez - Goa. -

Krishna Rebel Women
Krishna’s Life and Women Rebels Shri Krishna is one of the most amazing personality in Indian mythology and Indian Philosophy He has many unique facets • Son • Lover • Friend • Philosopher • Teacher • Savior • Husband • God Each one with its great attributes In this article we will see how Krishna is a reason for Rebel for many women on Indian History Radha’s Rebel against the societal norms “Ra” in Sanskrit means a call and “Dha” means one who runs Radha’s name itself talks about rebel- When Krishna calls her she runs to him According to some scripts Radha was already married, still she is considered as prime lover of Krishna, so much so that her name is taken before Krishna’s Name in some communities. They salute each other with a phrase “Radhe- Krishna” She is one who breaks all social norms by leaving her marriage Krishna-Radha love story is a metaphor for divine-human relationship, where Radha is the human devotee or soul who is frustrated with the past, obligations to social expectations and the ideas she inherited, who then longs for real meaning, the true love, the divine (Krishna). Rukmini and Her Rebel Against Her Own People Vidarbha was one of the ancient and powerful Kingdoms during Mahabharata. The ruler of this place was Bhishmak. Bhishmak had 5 sons and 1 daughter. Eldest among them was Rukmi and the name of princess was Rukmini. Rukmi wanted Rukmini to get married to Chedi King Shishupal, a strong enemy of Shri Krishna. The Strategy of Jarasandh, Shishupal and Rukmi was to encircle the new kingdom of Shri Krihna- Dwaraka and catch the trade advantages of both Mathura and Dwaraka.