Gene Networks and Modules in Atherosclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -
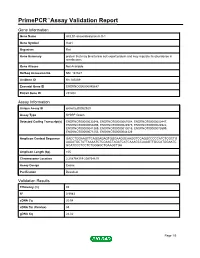
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name HCLS1-associated protein X-1 Gene Symbol Hax1 Organism Rat Gene Summary protein that may bind to bile salt export protein and may regulate its abundance in membranes Gene Aliases Not Available RefSeq Accession No. NM_181627 UniGene ID Rn.185269 Ensembl Gene ID ENSRNOG00000045647 Entrez Gene ID 291202 Assay Information Unique Assay ID qRnoCED0052920 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000030348, ENSRNOT00000067004, ENSRNOT00000000447, ENSRNOT00000059496, ENSRNOT00000024975, ENSRNOT00000024922, ENSRNOT00000041389, ENSRNOT00000010015, ENSRNOT00000073599, ENSRNOT00000071253, ENSRNOT00000044326 Amplicon Context Sequence GACCTGGAAGTTCAGGAGAGTGGGAAGGCAAGGTCCAGGCCCCCATCTCGCTG AAGATGCTATTAAAATCTCGAACTAGATCATCAAAGCCAAAGTTGCCATGGAATC GCATCCCTCCTCTGGGGCTGAAGCTGA Amplicon Length (bp) 105 Chromosome Location 2:208764319-208764619 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 92 R2 0.9983 cDNA Cq 20.54 cDNA Tm (Celsius) 83 gDNA Cq 24.02 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report Hax1, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Duke University Dissertation Template
Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 i v ABSTRACT Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 Copyright by Cavin Keith Ward-Caviness 2014 Abstract In this manuscript I seek to demonstrate the importance of gene-environment interactions in cardiovascular disease. This manuscript contains five studies each of which contributes to our understanding of the joint impact of genetic variation -

Identification of Expression Qtls Targeting Candidate Genes For
ISSN: 2378-3648 Salleh et al. J Genet Genome Res 2018, 5:035 DOI: 10.23937/2378-3648/1410035 Volume 5 | Issue 1 Journal of Open Access Genetics and Genome Research RESEARCH ARTICLE Identification of Expression QTLs Targeting Candidate Genes for Residual Feed Intake in Dairy Cattle Using Systems Genomics Salleh MS1,2, Mazzoni G2, Nielsen MO1, Løvendahl P3 and Kadarmideen HN2,4* 1Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark Check for 2Department of Bio and Health Informatics, Technical University of Denmark, Lyngby, Denmark updates 3Department of Molecular Biology and Genetics-Center for Quantitative Genetics and Genomics, Aarhus University, AU Foulum, Tjele, Denmark 4Department of Applied Mathematics and Computer Science, Technical University of Denmark, Lyngby, Denmark *Corresponding author: Kadarmideen HN, Department of Applied Mathematics and Computer Science, Technical University of Denmark, DK-2800, Kgs. Lyngby, Denmark, E-mail: [email protected] Abstract body weight gain and net merit). The eQTLs and biological pathways identified in this study improve our understanding Background: Residual feed intake (RFI) is the difference of the complex biological and genetic mechanisms that de- between actual and predicted feed intake and an important termine FE traits in dairy cattle. The identified eQTLs/genet- factor determining feed efficiency (FE). Recently, 170 can- ic variants can potentially be used in new genomic selection didate genes were associated with RFI, but no expression methods that include biological/functional information on quantitative trait loci (eQTL) mapping has hitherto been per- SNPs. formed on FE related genes in dairy cows. In this study, an integrative systems genetics approach was applied to map Keywords eQTLs in Holstein and Jersey cows fed two different diets to eQTL, RNA-seq, Genotype, Data integration, Systems improve identification of candidate genes for FE. -

Anti-Caspase 3 Antibody Catalog # ABO11880
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Anti-Caspase 3 Antibody Catalog # ABO11880 Specification Anti-Caspase 3 Antibody - Product Information Application WB, IHC Primary Accession P42574 Host Rabbit Reactivity Human, Mouse, Rat Clonality Polyclonal Format Lyophilized Description Rabbit IgG polyclonal antibody for Caspase-3(CASP3) detection. Tested with WB, IHC-P in Human;Mouse;Rat. Reconstitution Add 0.2ml of distilled water will yield a Anti- Caspase 3 antibody, ABO11880, concentration of 500ug/ml. Western blottingAll lanes: Anti Caspase 3 (ABO11880) at 0.5ug/mlWB: Recombinant Human Caspase 3 Protein 0.5ngPredicted Anti-Caspase 3 Antibody - Additional Information bind size: 39KDObserved bind size: 39KD Gene ID 836 Other Names Caspase-3, CASP-3, 3.4.22.56, Apopain, Cysteine protease CPP32, CPP-32, Protein Yama, SREBP cleavage activity 1, SCA-1, Caspase-3 subunit p17, Caspase-3 subunit p12, CASP3, CPP32 Calculated MW 31608 MW KDa Application Details Immunohistochemistry(Paraffin-embedded Section), 0.5-1 µg/ml, Human, Mouse, Rat, By Heat<br> <br>Western blot, 0.1-0.5 µg/ml, Human, Rat<br> Anti-Caspase 3 antibody, ABO11880, IHC(P)IHC(P): Human Lung Cancer Tissue Subcellular Localization Cytoplasm. Tissue Specificity Highly expressed in lung, spleen, heart, liver and kidney. Moderate levels in brain and skeletal muscle, and low in testis. Also found in many cell lines, highest expression in cells of the immune system. Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Protein Name Caspase-3 Contents Each vial contains 5mg BSA, 0.9mg NaCl, 0.2mg Na2HPO4, 0.05mg NaN3. -

WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12N 1/19 (2006.01) C12Q 1/02 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C12N 15/81 (2006.01) C07K 14/47 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 15/049674 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 11 September 2015 ( 11.09.201 5) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/050,045 12 September 2014 (12.09.2014) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: WHITEHEAD INSTITUTE FOR BIOMED¬ DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, ICAL RESEARCH [US/US]; Nine Cambridge Center, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Cambridge, Massachusetts 02142-1479 (US). -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Rabbit Anti-HCLS1/FITC Conjugated Antibody
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-HCLS1/FITC Conjugated antibody SL5109R-FITC Product Name: Anti-HCLS1/FITC Chinese Name: FITC标记的造血Stem cells特异性蛋白抗体 Cortactin like; CTTNL; HCLS 1; HCLS 1 protein; Hcls1; HCLS1 protein; HCLS1_HUMAN; Hematopoietic cell specific Lyn substrate 1; Hematopoietic cell- Alias: specific LYN substrate 1; Hematopoietic lineage cell specific protein; Hematopoietic lineage cell-specific protein; HS 1; HS1; LckBP 1; LckBP1; p75 antibody. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Cow,Horse,Rabbit, IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 53kDa Cellular localization: The cell membrane Form: Lyophilized or Liquid Concentration: 1mg/ml immunogen: KLH conjugated synthetic peptide derived from human HCLS1 Lsotype: IgGwww.sunlongbiotech.com Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. background: Substrate of the antigen receptor-coupled tyrosine kinase. Plays a role in antigen receptor signaling for both clonal expansion and deletion in lymphoid cells. May also Product Detail: be involved in the regulation of gene expression. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

HS1BP3 (N-17): Sc-161716
SAN TA C RUZ BI OTEC HNOL OG Y, INC . HS1BP3 (N-17): sc-161716 BACKGROUND APPLICATIONS HS1BP3 (HCLS1-binding protein 3), also known as ETM2 or HS1-BP3, is a 392 HS1BP3 (N-17) is recommended for detection of HS1BP3 of mouse, rat and amino acid protein that contains one PX (phox homology) domain, a leucine human origin by Western Blotting (starting dilution 1:200, dilution range zipper, immunoreceptor tyrosine-based inhibitory motif-like motifs and multi - 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein ple proline-rich regions. Expressed primarily in brain, HS1BP3 is encoded by (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution a gene mapping to human chromosome 2p24.1. The gene encoding HS1BP3 range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution is frequently mutated in familial essential tremor, a disorder characterized by range 1:30-1:3000). kinetic tremor the the hands, voice or head, though there is no correlation to HS1BP3 (N-17) is also recommended for detection of HS1BP3 in additional Parkinson disease. HS1BP3 interacts with HAX-1’s SH3 domain, and may also species, including equine, canine, bovine and porcine. play a role in the regulation of catecholamine and serotonin metabolism. Acting as a regulator of IL-2 signaling, HS1BP3 is likely involved in lympho cyte Suitable for use as control antibody for HS1BP3 siRNA (h): sc-94437, HS1BP3 activation. siRNA (m): sc-146083, HS1BP3 shRNA Plasmid (h): sc-94437-SH, HS1BP3 shRNA Plasmid (m): sc-146083-SH, HS1BP3 shRNA (h) Lentiviral Particles: REFERENCES sc- 94437-V and HS1BP3 shRNA (m) Lentiviral Particles: sc-146083-V. -
Sheet1 Page 1 Gene Symbol Gene Description Entrez Gene ID
Sheet1 RefSeq ID ProbeSets Gene Symbol Gene Description Entrez Gene ID Sequence annotation Seed matches location(s) Ago-2 binding specific enrichment (replicate 1) Ago-2 binding specific enrichment (replicate 2) OE lysate log2 fold change (replicate 1) OE lysate log2 fold change (replicate 2) Probability Pulled down in Karginov? NM_005646 202813_at TARBP1 Homo sapiens TAR (HIV-1) RNA binding protein 1 (TARBP1), mRNA. 6894 TR(1..5130)CDS(1..4866) 4868..4874,5006..5013 3.73 2.53 -1.54 -0.44 1 Yes NM_001665 203175_at RHOG Homo sapiens ras homolog gene family, member G (rho G) (RHOG), mRNA. 391 TR(1..1332)CDS(159..734) 810..817,782..788,790..796,873..879 3.56 2.78 -1.62 -1 1 Yes NM_002742 205880_at PRKD1 Homo sapiens protein kinase D1 (PRKD1), mRNA. 5587 TR(1..3679)CDS(182..2920) 3538..3544,3202..3208 4.15 1.83 -2.55 -0.42 1 Yes NM_003068 213139_at SNAI2 Homo sapiens snail homolog 2 (Drosophila) (SNAI2), mRNA. 6591 TR(1..2101)CDS(165..971) 1410..1417,1814..1820,1610..1616 3.5 2.79 -1.38 -0.31 1 Yes NM_006270 212647_at RRAS Homo sapiens related RAS viral (r-ras) oncogene homolog (RRAS), mRNA. 6237 TR(1..1013)CDS(46..702) 871..877 3.82 2.27 -1.54 -0.55 1 Yes NM_025188 219923_at,242056_at TRIM45 Homo sapiens tripartite motif-containing 45 (TRIM45), mRNA. 80263 TR(1..3584)CDS(589..2331) 3408..3414,2437..2444,3425..3431,2781..2787 3.87 1.89 -0.62 -0.09 1 Yes NM_024684 221600_s_at,221599_at C11orf67 Homo sapiens chromosome 11 open reading frame 67 (C11orf67), mRNA. -
Anti-Caspase-3 Polyclonal Antibody Description Product
9853 Pacific Heights Blvd. Suite D. San Diego, CA 92121, USA Tel: 858-263-4982 Email: [email protected] 39-2098: Anti-Caspase-3 Polyclonal Antibody Clonality : Polyclonal Application : WB Reactivity : Human Gene : CASP3 Gene ID : 836 Uniprot ID : P42574 Caspase-3; CASP-3; 3.4.22.56; Apopain; Cysteine protease CPP32; CPP-32; Protein Yama; SREBP Alternative Name : cleavage activity 1; SCA-1; Caspase-3 subunit p17; Caspase-3 subunit p12; CASP3; CPP32 Isotype : Rabbit IgG Description Caspase 3 is a caspase protein which interacts with Survivin, XIAP, CFLAR, Caspase 8, HCLS1, Deleted in Colorectal Cancer, TRAF3 and GroEL. This gene which is located at 4q35 encodes a protein that is a member of the cysteine-aspartic acid protease(caspase) family. Sequential activation of caspases plays a central role in the execution-phase of cell apoptosis. Caspases exist as inactive proenzymes that undergo proteolytic processing at conserved aspartic residues to produce two subunits, large and small, that dimerize to form the active enzyme. This protein cleaves and activates caspases 6, 7, and 9; and the protein itself is processed by caspases 8, 9, and 10. It is the predominant caspase involved in the cleavage of amyloid-beta 4A precursor protein, which is associated with neuronal death in Alzheimer's disease. And the caspase-3 activation in heart failure sequentially cleaves SRF and generates a truncated SRF that appears to function as a dominant-negative transcription fac Product Info Amount : 100 µg/vial Purification : Immunogen affinity purified. Each vial contains 5mg BSA, 0.9mg NaCl, 0.2mg Na2HPO4, 0.05mg Thimerosal, 0.05mg NaN3.