Identification of Loci Associated with Susceptibility to Bovine Paratuberculosis and with the Dysregulation of the MECOM, Eef1a2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Genome-Wide Association Study Identifies Loci for Arterial Stiffness
www.nature.com/scientificreports OPEN Genome-wide association study identifes loci for arterial stifness index in 127,121 UK Biobank Received: 1 February 2019 Accepted: 5 June 2019 participants Published: xx xx xxxx Kenneth Fung1, Julia Ramírez 2, Helen R. Warren2,3, Nay Aung 1, Aaron M. Lee1, Evan Tzanis2,3, Stefen E. Petersen 1,3 & Patricia B. Munroe2,3 Arterial stifness index (ASI) is a non-invasive measure of arterial stifness using infra-red fnger sensors (photoplethysmography). It is a well-suited measure for large populations as it is relatively inexpensive to perform, and data can be acquired within seconds. These features raise interest in using ASI as a tool to estimate cardiovascular disease risk as prior work demonstrates increased arterial stifness is associated with elevated systolic blood pressure, and ASI is predictive of cardiovascular disease and mortality. We conducted genome-wide association studies (GWASs) for ASI in 127,121 UK Biobank participants of European-ancestry. Our primary analyses identifed variants at four loci reaching genome-wide signifcance (P < 5 × 10−8): TEX41 (rs1006923; P = 5.3 × 10−12), FOXO1 (rs7331212; P = 2.2 × 10−11), C1orf21 (rs1930290, P = 1.1 × 10−8) and MRVI1 (rs10840457, P = 3.4 × 10−8). Gene- based testing revealed three signifcant genes, the most signifcant gene was COL4A2 (P = 1.41 × 10−8) encoding type IV collagen. Other candidate genes at associated loci were also involved in smooth muscle tone regulation. Our fndings provide new information for understanding the development of arterial stifness. Arterial stifness measures have been reported as independent markers of vascular ageing1,2, hypertension3,4, car- diovascular disease (CVD)5,6 and mortality6,7. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -
Primepcr™Assay Validation Report
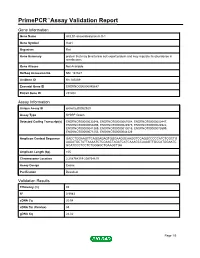
PrimePCR™Assay Validation Report Gene Information Gene Name HCLS1-associated protein X-1 Gene Symbol Hax1 Organism Rat Gene Summary protein that may bind to bile salt export protein and may regulate its abundance in membranes Gene Aliases Not Available RefSeq Accession No. NM_181627 UniGene ID Rn.185269 Ensembl Gene ID ENSRNOG00000045647 Entrez Gene ID 291202 Assay Information Unique Assay ID qRnoCED0052920 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000030348, ENSRNOT00000067004, ENSRNOT00000000447, ENSRNOT00000059496, ENSRNOT00000024975, ENSRNOT00000024922, ENSRNOT00000041389, ENSRNOT00000010015, ENSRNOT00000073599, ENSRNOT00000071253, ENSRNOT00000044326 Amplicon Context Sequence GACCTGGAAGTTCAGGAGAGTGGGAAGGCAAGGTCCAGGCCCCCATCTCGCTG AAGATGCTATTAAAATCTCGAACTAGATCATCAAAGCCAAAGTTGCCATGGAATC GCATCCCTCCTCTGGGGCTGAAGCTGA Amplicon Length (bp) 105 Chromosome Location 2:208764319-208764619 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 92 R2 0.9983 cDNA Cq 20.54 cDNA Tm (Celsius) 83 gDNA Cq 24.02 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report Hax1, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Impaired Iloprost-Induced Platelet Inhibition and Phosphoproteome Changes in Patients with Confirmed Pseudohypoparathyroidism Ty
www.nature.com/scientificreports OPEN Impaired iloprost‑induced platelet inhibition and phosphoproteome changes in patients with confrmed pseudohypoparathyroidism type Ia, linked to genetic mutations in GNAS Frauke Swieringa1,2, Fiorella A. Solari1,9, Oliver Pagel1,9, Florian Beck1, Jingnan Huang1,2, Marion A. H. Feijge2, Kerstin Jurk4, Irene M. L. W. Körver‑Keularts5, Nadine J. A. Mattheij2, Jörg Faber3, Joachim Pohlenz3, Alexandra Russo3, Connie T. R. M. Stumpel5,6, Dirk E. Schrander7, Barbara Zieger8, Paola E. J. van der Meijden2, René P. Zahedi1, Albert Sickmann1 & Johan W. M. Heemskerk2* Patients diagnosed with pseudohypoparathyroidism type Ia (PHP Ia) sufer from hormonal resistance and abnormal postural features, in a condition classifed as Albright hereditary osteodystrophy (AHO) syndrome. This syndrome is linked to a maternally inherited mutation in the GNAS complex locus, encoding for the GTPase subunit Gsα. Here, we investigated how platelet phenotype and omics analysis can assist in the often difcult diagnosis. By coupling to the IP receptor, Gsα induces platelet inhibition via adenylyl cyclase and cAMP-dependent protein kinase A (PKA). In platelets from seven patients with suspected AHO, one of the largest cohorts examined, we studied the PKA-induced phenotypic changes. Five patients with a confrmed GNAS mutation, displayed impairments in Gsα- dependent VASP phosphorylation, aggregation, and microfuidic thrombus formation. Analysis of the platelet phosphoproteome revealed 2,516 phosphorylation sites, of which 453 were regulated by Gsα-PKA. Common changes in the patients were: (1) a joint panel of upregulated and downregulated phosphopeptides; (2) overall PKA dependency of the upregulated phosphopeptides; (3) links to key platelet function pathways. In one patient with GNAS mutation, diagnosed as non‑AHO, the changes in platelet phosphoproteome were reversed. -

Duke University Dissertation Template
Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 i v ABSTRACT Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 Copyright by Cavin Keith Ward-Caviness 2014 Abstract In this manuscript I seek to demonstrate the importance of gene-environment interactions in cardiovascular disease. This manuscript contains five studies each of which contributes to our understanding of the joint impact of genetic variation -

Identification of Expression Qtls Targeting Candidate Genes For
ISSN: 2378-3648 Salleh et al. J Genet Genome Res 2018, 5:035 DOI: 10.23937/2378-3648/1410035 Volume 5 | Issue 1 Journal of Open Access Genetics and Genome Research RESEARCH ARTICLE Identification of Expression QTLs Targeting Candidate Genes for Residual Feed Intake in Dairy Cattle Using Systems Genomics Salleh MS1,2, Mazzoni G2, Nielsen MO1, Løvendahl P3 and Kadarmideen HN2,4* 1Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark Check for 2Department of Bio and Health Informatics, Technical University of Denmark, Lyngby, Denmark updates 3Department of Molecular Biology and Genetics-Center for Quantitative Genetics and Genomics, Aarhus University, AU Foulum, Tjele, Denmark 4Department of Applied Mathematics and Computer Science, Technical University of Denmark, Lyngby, Denmark *Corresponding author: Kadarmideen HN, Department of Applied Mathematics and Computer Science, Technical University of Denmark, DK-2800, Kgs. Lyngby, Denmark, E-mail: [email protected] Abstract body weight gain and net merit). The eQTLs and biological pathways identified in this study improve our understanding Background: Residual feed intake (RFI) is the difference of the complex biological and genetic mechanisms that de- between actual and predicted feed intake and an important termine FE traits in dairy cattle. The identified eQTLs/genet- factor determining feed efficiency (FE). Recently, 170 can- ic variants can potentially be used in new genomic selection didate genes were associated with RFI, but no expression methods that include biological/functional information on quantitative trait loci (eQTL) mapping has hitherto been per- SNPs. formed on FE related genes in dairy cows. In this study, an integrative systems genetics approach was applied to map Keywords eQTLs in Holstein and Jersey cows fed two different diets to eQTL, RNA-seq, Genotype, Data integration, Systems improve identification of candidate genes for FE. -
Primepcr™Assay Validation Report
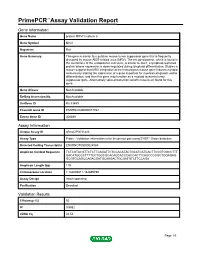
PrimePCR™Assay Validation Report Gene Information Gene Name protein MRVI1 isoform b Gene Symbol Mrvi1 Organism Rat Gene Summary This gene is similar to a putative mouse tumor suppressor gene that is frequently disrupted by mouse AIDS-related virus (MRV). The encoded protein, which is found in the membrane of the endoplasmic reticulum, is similar to Jaw1, a lymphoid-restricted protein whose expression is down-regulated during lymphoid differentiation. Studies in mouse suggest that MRV integration at the homologous mouse gene induces myeloid leukemia by altering the expression of a gene important for myeloid cell growth and/or differentiation, and thus this gene may function as a myeloid leukemia tumor suppressor gene. Alternatively spliced transcript variants have been found for this gene. Gene Aliases Not Available RefSeq Accession No. Not Available UniGene ID Rn.33889 Ensembl Gene ID ENSRNOG00000017767 Entrez Gene ID 308899 Assay Information Unique Assay ID qRnoCIP0031443 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000024060 Amplicon Context Sequence TCTCGTACGTTCTCTTCAGATTCTCCACATACTGCATCATCACTTCCGTGGCCTTT GACATGCGCTTTTCCTGGCGCACAGCACCCACCACTTCAGCCCGGCTGGAGAG GCGTGCAGCCAGACGATGCAAGACTGCGATGTCTTCCAGA Amplicon Length (bp) 119 Chromosome Location 1:182696811-182699799 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 95 R2 0.9982 cDNA Cq 24.54 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 88 gDNA Cq Specificity -

Mrvil, a Common MRV Integration Site in BXH2 Myeloid Leukemias, Encodes a Protein with Homology to a Lymphoid-Restricted Membrane Protein Jaw1
Oncogene (1999) 18, 2069 ± 2084 ã 1999 Stockton Press All rights reserved 0950 ± 9232/99 $12.00 http://www.stockton-press.co.uk/onc Mrvil, a common MRV integration site in BXH2 myeloid leukemias, encodes a protein with homology to a lymphoid-restricted membrane protein Jaw1 John D Shaughnessy Jr*,1, David A Largaespada2, Erming Tian1, Colin F Fletcher3, Brian C Cho4, Paresh Vyas5, Nancy A Jenkins3 and Neal G Copeland3 1Division of Hematology and Oncology, Department of Medicine, University of Arkansas for Medical Sciences, Little Rock, Arkansas, AR 72205, USA; 2Department of Laboratory Medicine and Pathology, University of Minnesota Cancer Center, Minneapolis, Minnesota, MN 55455, USA; 3Mammalian Genetics Laboratory, ABL-Basic Research Program, NCI-Frederick Cancer Research and Development Center, Frederick, Maryland, MD 21702, USA; 4Department of Microbiology and Immunology, Thomas Jeerson University Cancer Institute, Philadelphia, Pennsylvania, PA 19107, USA; 5Department of Hematology and Oncology, Children's Hospital, Boston, Massachusetts, USA Ecotropic MuLVs induce myeloid leukemia in BXH2 Introduction mice by insertional mutagenesis of cellular proto- oncogenes or tumor suppressor genes. Disease genes BXH2 mice represent an important model for the can thus be identi®ed by viral tagging as common sites of identi®cation of myeloid leukemia disease genes. Not viral integration in BXH2 leukemias. Previous studies only do these mice have the highest spontaneous showed that a frequent common integration site in BXH2 incidence of myeloid leukemia of any known inbred leukemias is the Nf1 tumor suppressor gene. Unexpect- mouse strain, but the leukemias in these mice are edly, about half of the viral integrations at Nf1 retrovirally-induced and the proviruses in the tumors represented a previously undiscovered defective none- can thus be used as insertional tags to identify the cotropic virus, termed MRV. -

Description: Uniprot:Q13976 Alternative Names: Specificity
TD2677 PRKG1 Antibody Order 021-34695924 [email protected] Support 400-6123-828 50ul [email protected] 100 uL √ √ Web www.ab-mart.com.cn Description: Serine/threonine protein kinase that acts as key mediator of the nitric oxide (NO)/cGMP signaling pathway. GMP binding activates PRKG1, which phosphorylates serines and threonines on many cellular proteins. Numerous protein targets for PRKG1 phosphorylation are implicated in modulating cellular calcium, but the contribution of each of these targets may vary substantially among cell types. Proteins that are phosphorylated by PRKG1 regulate platelet activation and adhesion, smooth muscle contraction, cardiac function, gene expression, feedback of the NO-signaling pathway, and other processes involved in several aspects of the CNS like axon guidance, hippocampal and cerebellar learning, circadian rhythm and nociception. Smooth muscle relaxation is mediated through lowering of intracellular free calcium, by desensitization of contractile proteins to calcium, and by decrease in the contractile state of smooth muscle or in platelet activation. Regulates intracellular calcium levels via several pathways: phosphorylates MRVI1/IRAG and inhibits IP3-induced Ca(2+) release from intracellular stores, phosphorylation of KCNMA1 (BKCa) channels decreases intracellular Ca(2+) levels, which leads to increased opening of this channel. PRKG1 phosphorylates the canonical transient receptor potential channel (TRPC) family which inactivates the associated inward calcium current. Another mode of action of NO/cGMP/PKGI signaling involves PKGI-mediated inactivation of the Ras homolog gene family member A (RhoA). Phosphorylation of RHOA by PRKG1 blocks the action of this protein in myriad processes: regulation of RHOA translocation; decreasing contraction; controlling vesicle trafficking, reduction of myosin light chain phosphorylation resulting in vasorelaxation. -

Anti-Caspase 3 Antibody Catalog # ABO11880
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Anti-Caspase 3 Antibody Catalog # ABO11880 Specification Anti-Caspase 3 Antibody - Product Information Application WB, IHC Primary Accession P42574 Host Rabbit Reactivity Human, Mouse, Rat Clonality Polyclonal Format Lyophilized Description Rabbit IgG polyclonal antibody for Caspase-3(CASP3) detection. Tested with WB, IHC-P in Human;Mouse;Rat. Reconstitution Add 0.2ml of distilled water will yield a Anti- Caspase 3 antibody, ABO11880, concentration of 500ug/ml. Western blottingAll lanes: Anti Caspase 3 (ABO11880) at 0.5ug/mlWB: Recombinant Human Caspase 3 Protein 0.5ngPredicted Anti-Caspase 3 Antibody - Additional Information bind size: 39KDObserved bind size: 39KD Gene ID 836 Other Names Caspase-3, CASP-3, 3.4.22.56, Apopain, Cysteine protease CPP32, CPP-32, Protein Yama, SREBP cleavage activity 1, SCA-1, Caspase-3 subunit p17, Caspase-3 subunit p12, CASP3, CPP32 Calculated MW 31608 MW KDa Application Details Immunohistochemistry(Paraffin-embedded Section), 0.5-1 µg/ml, Human, Mouse, Rat, By Heat<br> <br>Western blot, 0.1-0.5 µg/ml, Human, Rat<br> Anti-Caspase 3 antibody, ABO11880, IHC(P)IHC(P): Human Lung Cancer Tissue Subcellular Localization Cytoplasm. Tissue Specificity Highly expressed in lung, spleen, heart, liver and kidney. Moderate levels in brain and skeletal muscle, and low in testis. Also found in many cell lines, highest expression in cells of the immune system. Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Protein Name Caspase-3 Contents Each vial contains 5mg BSA, 0.9mg NaCl, 0.2mg Na2HPO4, 0.05mg NaN3. -

WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/040794 Al 17 March 2016 (17.03.2016) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12N 1/19 (2006.01) C12Q 1/02 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C12N 15/81 (2006.01) C07K 14/47 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 15/049674 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 11 September 2015 ( 11.09.201 5) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/050,045 12 September 2014 (12.09.2014) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: WHITEHEAD INSTITUTE FOR BIOMED¬ DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, ICAL RESEARCH [US/US]; Nine Cambridge Center, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Cambridge, Massachusetts 02142-1479 (US).