(Q13;Q12) Translocation Fusing FLT3 with GOLGB1: Toward Myeloid/Lymphoid Neoplasms with Eosinophilia and Rearrangement of FLT3?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Novel 65-Bp Indel in the GOLGB1 Gene Is Associated with Chicken Growth and Carcass Traits
animals Article A Novel 65-bp Indel in the GOLGB1 Gene Is Associated with Chicken Growth and Carcass Traits Rong Fu 1,2, Tuanhui Ren 1,2, Wangyu Li 1,2, Jiaying Liang 1,2, Guodong Mo 1,2, Wen Luo 1,2, Danlin He 1,2, Shaodong Liang 1,2 and Xiquan Zhang 1,2,* 1 Department of Animal Genetics, Breeding and Reproduction, College of Animal Science, South China Agricultural University, Guangzhou 510642, China; [email protected] (R.F.); [email protected] (T.R.); [email protected] (W.L.); [email protected] (J.L.); [email protected] (G.M.); [email protected] (W.L.); [email protected] (D.H.); [email protected] (S.L.) 2 Guangdong Provincial Key Lab of Agro-Animal Genomics and Molecular Breeding, and Key Laboratory of Chicken Genetics, Breeding and Reproduction, Ministry of Agriculture, Guangzhou 510642, China * Correspondence: [email protected] Received: 20 February 2020; Accepted: 4 March 2020; Published: 12 March 2020 Simple Summary: Many Chinese-local chickens show slow-growing and low-producing performance, which is not conductive to the development of the poultry industry. The identification of thousands of indels in the last twenty years has helped us to make progress in animal genetics and breeding. Golgin subfamily B member 1 (GOLGB1) is located on chromosome 1 in chickens. Previous study showed that a large number of QTLs on the chicken chromosome 1 were related to the important economic traits. However, the biological function of GOLGB1 gene in chickens is still unclear. In this study, we detected a novel 65-bp indel in the fifth intron of the chicken GOLGB1 gene. -
![Viewed Previously [4]](https://docslib.b-cdn.net/cover/6213/viewed-previously-4-126213.webp)
Viewed Previously [4]
Barlow et al. BMC Biology (2018) 16:27 https://doi.org/10.1186/s12915-018-0492-9 RESEARCHARTICLE Open Access A sophisticated, differentiated Golgi in the ancestor of eukaryotes Lael D. Barlow1, Eva Nývltová2,3, Maria Aguilar1, Jan Tachezy2 and Joel B. Dacks1,4* Abstract Background: The Golgi apparatus is a central meeting point for the endocytic and exocytic systems in eukaryotic cells, and the organelle’s dysfunction results in human disease. Its characteristic morphology of multiple differentiated compartments organized into stacked flattened cisternae is one of the most recognizable features of modern eukaryotic cells, and yet how this is maintained is not well understood. The Golgi is also an ancient aspect of eukaryotes, but the extent and nature of its complexity in the ancestor of eukaryotes is unclear. Various proteins have roles in organizing the Golgi, chief among them being the golgins. Results: We address Golgi evolution by analyzing genome sequences from organisms which have lost stacked cisternae as a feature of their Golgi and those that have not. Using genomics and immunomicroscopy, we first identify Golgi in the anaerobic amoeba Mastigamoeba balamuthi. We then searched 87 genomes spanning eukaryotic diversity for presence of the most prominent proteins implicated in Golgi structure, focusing on golgins. We show some candidates as animal specific and others as ancestral to eukaryotes. Conclusions: None of the proteins examined show a phyletic distribution that correlates with the morphology of stacked cisternae, suggesting the possibility of stacking as an emergent property. Strikingly, however, the combination of golgins conserved among diverse eukaryotes allows for the most detailed reconstruction of the organelle to date, showing a sophisticated Golgi with differentiated compartments and trafficking pathways in the common eukaryotic ancestor. -

Knock-Out of ACBD3 Leads to Dispersed Golgi Structure, but Unaffected Mitochondrial Functions in HEK293 and Hela Cells
International Journal of Molecular Sciences Article Knock-Out of ACBD3 Leads to Dispersed Golgi Structure, but Unaffected Mitochondrial Functions in HEK293 and HeLa Cells Tereza Da ˇnhelovská 1 , Lucie Zdražilová 1 , Hana Štufková 1, Marie Vanišová 1, Nikol Volfová 1, Jana Kˇrížová 1 , OndˇrejKuda 2 , Jana Sládková 1 and Markéta Tesaˇrová 1,* 1 Department of Paediatrics and Inherited Metabolic Disorders, Charles University, First Faculty of Medicine and General University Hospital in Prague, 128 01 Prague, Czech Republic; [email protected] (T.D.); [email protected] (L.Z.); [email protected] (H.Š.); [email protected] (M.V.); [email protected] (N.V.); [email protected] (J.K.); [email protected] (J.S.) 2 Institute of Physiology, Academy of Sciences of the Czech Republic, 142 00 Prague, Czech Republic; [email protected] * Correspondence: [email protected] Abstract: The Acyl-CoA-binding domain-containing protein (ACBD3) plays multiple roles across the cell. Although generally associated with the Golgi apparatus, it operates also in mitochondria. In steroidogenic cells, ACBD3 is an important part of a multiprotein complex transporting cholesterol into mitochondria. Balance in mitochondrial cholesterol is essential for proper mitochondrial protein biosynthesis, among others. We generated ACBD3 knock-out (ACBD3-KO) HEK293 and HeLa cells and characterized the impact of protein absence on mitochondria, Golgi, and lipid profile. In ACBD3- Citation: Daˇnhelovská,T.; KO cells, cholesterol level and mitochondrial structure and functions are not altered, demonstrating Zdražilová, L.; Štufková, H.; that an alternative pathway of cholesterol transport into mitochondria exists. However, ACBD3- Vanišová, M.; Volfová, N.; Kˇrížová,J.; Kuda, O.; Sládková, J.; Tesaˇrová,M. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Signal Peptide Peptidase‐Like 2C Impairs Vesicular Transport And
Article Signal peptide peptidase-like 2c impairs vesicular transport and cleaves SNARE proteins Alkmini A Papadopoulou1, Stephan A Müller2, Torben Mentrup3, Merav D Shmueli2,4,5, Johannes Niemeyer3, Martina Haug-Kröper1, Julia von Blume6, Artur Mayerhofer7, Regina Feederle2,8,9 , Bernd Schröder3,10 , Stefan F Lichtenthaler2,5,9 & Regina Fluhrer1,2,* Abstract Introduction Members of the GxGD-type intramembrane aspartyl proteases The high degree of compartmentalization in eukaryotic cells creates have emerged as key players not only in fundamental cellular a need for specific and precise protein trafficking. To meet this need, processes such as B-cell development or protein glycosylation, but cells have developed a complex system of vesicle transport that also in development of pathologies, such as Alzheimer’s disease or ensures safe sorting of cargo proteins, in particular between the dif- hepatitis virus infections. However, one member of this protease ferent compartments of the secretory pathway [1–3]. Vesicles origi- family, signal peptide peptidase-like 2c (SPPL2c), remains orphan nate from a donor membrane, translocate in a targeted manner, and and its capability of proteolysis as well as its physiological function get specifically tethered to the target membrane, before fusing with is still enigmatic. Here, we demonstrate that SPPL2c is catalytically it. Soluble N-ethylmaleimide-sensitive factor attachment protein active and identify a variety of SPPL2c candidate substrates using receptor (SNARE) proteins are known since three decades to medi- proteomics. The majority of the SPPL2c candidate substrates clus- ate specific membrane fusion [4,5]. So far, 38 SNARE proteins have ter to the biological process of vesicular trafficking. -

The Murine Orthologue of the Golgi-Localized TPTE Protein Provides Clues to the Evolutionary History of the Human TPTE Gene Family
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Hum Genet (2001) 109:569–575 DOI 10.1007/s004390100607 ORIGINAL INVESTIGATION Michel Guipponi · Caroline Tapparel · Olivier Jousson · Nathalie Scamuffa · Christophe Mas · Colette Rossier · Pierre Hutter · Paolo Meda · Robert Lyle · Alexandre Reymond · Stylianos E. Antonarakis The murine orthologue of the Golgi-localized TPTE protein provides clues to the evolutionary history of the human TPTE gene family Received: 5 June 2001 / Accepted: 17 August 2001 / Published online: 27 October 2001 © Springer-Verlag 2001 Abstract The human TPTE gene encodes a testis-spe- events. The Y chromosome copy of TPTE is a pseudo- cific protein that contains four potential transmembrane gene and is not therefore involved in the testis expression domains and a protein tyrosine phosphatase motif, and of this gene family. shows homology to the tumor suppressor PTEN/MMAC1. Chromosomal mapping revealed multiple copies of the TPTE gene present on the acrocentric chromosomes 13, Introduction 15, 21 and 22, and the Y chromosome. Zooblot analysis suggests that mice may possess only one copy of TPTE. We have recently identified a testis-specific cDNA, TPTE In the present study, we report the isolation and initial (Transmembrane Phosphatase with TEnsin homology), characterization of the full-length cDNA of the mouse ho- that encodes a predicted protein of 551 amino acids con- mologue Tpte. At least three different mRNA transcripts taining four potential transmembrane domains and a tyro- (Tpte.a, b, c) are produced via alternative splicing, encod- sine phosphatase motif (Chen H et al. -

Behavioral and Other Phenotypes in a Cytoplasmic Dynein Light Intermediate Chain 1 Mutant Mouse
The Journal of Neuroscience, April 6, 2011 • 31(14):5483–5494 • 5483 Cellular/Molecular Behavioral and Other Phenotypes in a Cytoplasmic Dynein Light Intermediate Chain 1 Mutant Mouse Gareth T. Banks,1* Matilda A. Haas,5* Samantha Line,6 Hazel L. Shepherd,6 Mona AlQatari,7 Sammy Stewart,7 Ida Rishal,8 Amelia Philpott,9 Bernadett Kalmar,2 Anna Kuta,1 Michael Groves,3 Nicholas Parkinson,1 Abraham Acevedo-Arozena,10 Sebastian Brandner,3,4 David Bannerman,6 Linda Greensmith,2,4 Majid Hafezparast,9 Martin Koltzenburg,2,4,7 Robert Deacon,6 Mike Fainzilber,8 and Elizabeth M. C. Fisher1,4 1Department of Neurodegenerative Disease, 2Sobell Department of Motor Science and Movement Disorders, 3Division of Neuropathology, and 4Medical Research Council (MRC) Centre for Neuromuscular Diseases, University College London (UCL) Institute of Neurology, London WC1N 3BG, United Kingdom, 5MRC National Institute for Medical Research, London NW7 1AA, United Kingdom, 6Department of Experimental Psychology, University of Oxford, Oxford OX1 3UD, United Kingdom, 7UCL Institute of Child Health, London WC1N 1EH, United Kingdom, 8Department of Biological Chemistry, Weizmann Institute of Science, 76100 Rehovot, Israel, 9School of Life Sciences, University of Sussex, Brighton BN1 9QG, United Kingdom, and 10MRC Mammalian Genetics Unit, Harwell OX11 ORD, United Kingdom The cytoplasmic dynein complex is fundamentally important to all eukaryotic cells for transporting a variety of essential cargoes along microtubules within the cell. This complex also plays more specialized roles in neurons. The complex consists of 11 types of protein that interact with each other and with external adaptors, regulators and cargoes. Despite the importance of the cytoplasmic dynein complex, weknowcomparativelylittleoftherolesofeachcomponentprotein,andinmammalsfewmutantsexistthatallowustoexploretheeffects of defects in dynein-controlled processes in the context of the whole organism. -

Multifaceted Polo-Like Kinases: Drug Targets and Antitargets for Cancer Therapy
REVIEWS Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy Klaus Strebhardt Abstract | The polo-like kinase 1 (PLK1) acts in concert with cyclin-dependent kinase 1–cyclin B1 and Aurora kinases to orchestrate a wide range of critical cell cycle events. Because PLK1 has been preclinically validated as a cancer target, small-molecule inhibitors of PLK1 have become attractive candidates for anticancer drug development. Although the roles of the closely related PLK2, PLK3 and PLK4 in cancer are less well understood, there is evidence showing that PLK2 and PLK3 act as tumour suppressors through their functions in the p53 signalling network, which guards the cell against various stress signals. In this article, recent insights into the biology of PLKs will be reviewed, with an emphasis on their role in malignant transformation, and progress in the development of small-molecule PLK1 inhibitors will be examined. More than two decades ago polo, the founding member Early observations on the overexpression of PLK1 of the family of polo-like kinases (PLKs), was identified in human tumours12 and on the inhibition of cellu- as having an essential role in the ordered execution of lar proliferation by microinjecting antibodies against mitotic events in Drosophila melanogaster 1,2. Since the PLK1 into HeLa cells13 initiated a series of follow-on discovery of polo, a wealth of functional information on studies using a broad spectrum of inhibitors (for exam- PLKs has been collected in a wide phylogenetic space. ple, dominant-negative forms of PLK1, antisense oli- Five mammalian PLK family members have been iden- gonucleotides and small interfering RNAs) aiming to tified so far, PLK1 (also known as STPK13), PLK2 (also evaluate PLK1 as a potential target for the treatment known as SNK), PLK3 (also known as CNK, FNK and of cancer14–17. -
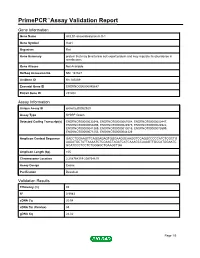
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name HCLS1-associated protein X-1 Gene Symbol Hax1 Organism Rat Gene Summary protein that may bind to bile salt export protein and may regulate its abundance in membranes Gene Aliases Not Available RefSeq Accession No. NM_181627 UniGene ID Rn.185269 Ensembl Gene ID ENSRNOG00000045647 Entrez Gene ID 291202 Assay Information Unique Assay ID qRnoCED0052920 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000030348, ENSRNOT00000067004, ENSRNOT00000000447, ENSRNOT00000059496, ENSRNOT00000024975, ENSRNOT00000024922, ENSRNOT00000041389, ENSRNOT00000010015, ENSRNOT00000073599, ENSRNOT00000071253, ENSRNOT00000044326 Amplicon Context Sequence GACCTGGAAGTTCAGGAGAGTGGGAAGGCAAGGTCCAGGCCCCCATCTCGCTG AAGATGCTATTAAAATCTCGAACTAGATCATCAAAGCCAAAGTTGCCATGGAATC GCATCCCTCCTCTGGGGCTGAAGCTGA Amplicon Length (bp) 105 Chromosome Location 2:208764319-208764619 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 92 R2 0.9983 cDNA Cq 20.54 cDNA Tm (Celsius) 83 gDNA Cq 24.02 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report Hax1, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Datasheet: MCA3923Z Product Details
Datasheet: MCA3923Z Description: MOUSE ANTI HUMAN ACBD3:Preservative Free Specificity: ACBD3 Format: Preservative Free Product Type: Monoclonal Antibody Clone: 2G2 Isotype: IgG1 Quantity: 0.1 mg Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Immunohistology - Paraffin (1) 0.1 - 10 ug/ml Western Blotting Immunofluorescence 0.1 - 10 ug/ml Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Suggested working dilutions are given as a guide only. It is recommended that the user titrates the product for use in their own system using appropriate negative/positive controls. (1)This product requires antigen retrieval using heat treatment prior to staining of paraffin sections.Sodium citrate buffer pH 6.0 is recommended for this purpose. Target Species Human Species Cross Reacts with: Rat, Mouse Reactivity N.B. Antibody reactivity and working conditions may vary between species. Product Form Purified IgG - liquid Preparation Purified IgG prepared by affinity chromatography on Protein A Buffer Solution Phosphate buffered saline Preservative None present Stabilisers Approx. Protein Ig concentration 0.5 mg/ml Concentrations Immunogen Recombinant protein corresponding to aa 73-172 of human ACBD3 Page 1 of 3 External Database Links UniProt: Q9H3P7 Related reagents Entrez Gene: 64746 ACBD3 Related reagents Synonyms GCP60, GOCAP1, GOLPH1 Fusion Partners Spleen cells from immunised Balb/c mice were fused with cells from the Sp2/0 myeloma cell line. -

Duke University Dissertation Template
Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 i v ABSTRACT Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 Copyright by Cavin Keith Ward-Caviness 2014 Abstract In this manuscript I seek to demonstrate the importance of gene-environment interactions in cardiovascular disease. This manuscript contains five studies each of which contributes to our understanding of the joint impact of genetic variation -

Identification of Expression Qtls Targeting Candidate Genes For
ISSN: 2378-3648 Salleh et al. J Genet Genome Res 2018, 5:035 DOI: 10.23937/2378-3648/1410035 Volume 5 | Issue 1 Journal of Open Access Genetics and Genome Research RESEARCH ARTICLE Identification of Expression QTLs Targeting Candidate Genes for Residual Feed Intake in Dairy Cattle Using Systems Genomics Salleh MS1,2, Mazzoni G2, Nielsen MO1, Løvendahl P3 and Kadarmideen HN2,4* 1Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark Check for 2Department of Bio and Health Informatics, Technical University of Denmark, Lyngby, Denmark updates 3Department of Molecular Biology and Genetics-Center for Quantitative Genetics and Genomics, Aarhus University, AU Foulum, Tjele, Denmark 4Department of Applied Mathematics and Computer Science, Technical University of Denmark, Lyngby, Denmark *Corresponding author: Kadarmideen HN, Department of Applied Mathematics and Computer Science, Technical University of Denmark, DK-2800, Kgs. Lyngby, Denmark, E-mail: [email protected] Abstract body weight gain and net merit). The eQTLs and biological pathways identified in this study improve our understanding Background: Residual feed intake (RFI) is the difference of the complex biological and genetic mechanisms that de- between actual and predicted feed intake and an important termine FE traits in dairy cattle. The identified eQTLs/genet- factor determining feed efficiency (FE). Recently, 170 can- ic variants can potentially be used in new genomic selection didate genes were associated with RFI, but no expression methods that include biological/functional information on quantitative trait loci (eQTL) mapping has hitherto been per- SNPs. formed on FE related genes in dairy cows. In this study, an integrative systems genetics approach was applied to map Keywords eQTLs in Holstein and Jersey cows fed two different diets to eQTL, RNA-seq, Genotype, Data integration, Systems improve identification of candidate genes for FE.