Cardiolipin Or MTCH2 Can Serve As Tbid Receptors During Apoptosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MARCH5 Requires MTCH2 to Coordinate Proteasomal Turnover of the MCL1:NOXA Complex
Cell Death & Differentiation (2020) 27:2484–2499 https://doi.org/10.1038/s41418-020-0517-0 ARTICLE MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1:NOXA complex 1,2 1,2,5 1,2 1,2 1,2,3 1,2 Tirta Mario Djajawi ● Lei Liu ● Jia-nan Gong ● Allan Shuai Huang ● Ming-jie Luo ● Zhen Xu ● 4 1,2 1,2 1,2 Toru Okamoto ● Melissa J. Call ● David C. S. Huang ● Mark F. van Delft Received: 3 July 2019 / Revised: 6 February 2020 / Accepted: 7 February 2020 / Published online: 24 February 2020 © The Author(s) 2020. This article is published with open access Abstract MCL1, a BCL2 relative, is critical for the survival of many cells. Its turnover is often tightly controlled through both ubiquitin-dependent and -independent mechanisms of proteasomal degradation. Several cell stress signals, including DNA damage and cell cycle arrest, are known to elicit distinct E3 ligases to ubiquitinate and degrade MCL1. Another trigger that drives MCL1 degradation is engagement by NOXA, one of its BH3-only protein ligands, but the mechanism responsible has remained unclear. From an unbiased genome-wide CRISPR-Cas9 screen, we discovered that the ubiquitin E3 ligase MARCH5, the ubiquitin E2 conjugating enzyme UBE2K, and the mitochondrial outer membrane protein MTCH2 co- — fi 1234567890();,: 1234567890();,: operate to mark MCL1 for degradation by the proteasome speci cally when MCL1 is engaged by NOXA. This mechanism of degradation also required the MCL1 transmembrane domain and distinct MCL1 lysine residues to proceed, suggesting that the components likely act on the MCL1:NOXA complex by associating with it in a specific orientation within the mitochondrial outer membrane. -

Cardiolipin and Mitochondrial Cristae Organization
Biochimica et Biophysica Acta 1859 (2017) 1156–1163 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamem Cardiolipin and mitochondrial cristae organization Nikita Ikon, Robert O. Ryan ⁎ Children's Hospital Oakland Research Institute, 5700 Martin Luther King Jr. Way, Oakland, CA 94609, United States article info abstract Article history: A fundamental question in cell biology, under investigation for over six decades, is the structural organization of Received 23 December 2016 mitochondrial cristae. Long known to harbor electron transport chain proteins, crista membrane integrity is key Received in revised form 3 March 2017 to establishment of the proton gradient that drives oxidative phosphorylation. Visualization of cristae morphol- Accepted 18 March 2017 ogy by electron microscopy/tomography has provided evidence that cristae are tube-like extensions of the mito- Available online 20 March 2017 chondrial inner membrane (IM) that project into the matrix space. Reconciling ultrastructural data with the lipid Keywords: composition of the IM provides support for a continuously curved cylindrical bilayer capped by a dome-shaped Cardiolipin tip. Strain imposed by the degree of curvature is relieved by an asymmetric distribution of phospholipids in Mitochondria monolayer leaflets that comprise cristae membranes. The signature mitochondrial lipid, cardiolipin (~18% of Cristae IM phospholipid mass), and phosphatidylethanolamine (34%) segregate to the negatively curved monolayer leaf- Membrane curvature let facing the crista lumen while the opposing, positively curved, matrix-facing monolayer leaflet contains pre- Non-bilayer lipid dominantly phosphatidylcholine. Associated with cristae are numerous proteins that function in distinctive Electron transport chain ways to establish and/or maintain their lipid repertoire and structural integrity. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Mechanisms Controlling Cell Life and Death Decisions
Department of Biological Regulation echanisms controlling cell Atan Gross Yehudit Zaltsman, Mlife and death decisions Natalie Yivgi-Ohana, Iris Kamer, Galia Oberkovitz, Liat Shachnai, Maria Maryanovich Programmed cell death or apoptosis in the extraembryonic (ExEm) region of is essential for both the development E7.5 wild type embryos, and this region and maintenance of tissue homeostasis is largely impaired in the Mtch2/Mimp-/- in multicellular organisms. The BCL-2 embryos. Thus, Mtch2/Mimp might play family members are critical regulators a critical role in the formation of the 972 8 934 3656 of the apoptotic program, whereas ExEm region. To study the connection the caspase proteases are the major between Mtch2/Mimp and apoptosis FAX 972 8 934 4116 executioners of this program. Members we generated Mtch2/Mimp-/- stable [email protected] of the BCL-2 family include both anti- and embryonic stem (ES) cell lines carrying www.weizmann.ac.il/ pro-apoptotic proteins. The BH3-only either an empty vector or Mtch2/Mimp. Biological_Regulation/gross proteins (e.g., BID) are an important Using these lines we demonstrated that subset of the pro-apoptotic proteins the presence of Mtch2/Mimp sensitizes that act as sentinels of intercellular cells to tBID-induced MOMP. Thus, we damage. In our laboratory, we are discovered that Mtch2/Mimp is critical S phase and inhibition of apoptosis focused on elucidating the mechanisms for normal embryonic development, following DNA damage. This surprising that balance between cell life and and is an important positive regulator and new pro-survival function of BID death, and BID is one of our primary of tBID-induced apoptosis at the is regulated by its phosphorylation tools to study these mechanisms. -

Serum from Pediatric Dilated Cardiomyopathy Patients Causes Dysregulation of Cardiolipin Biosynthesis and Mitochondrial Function Julie Pires Da Silva, Anastacia M
Serum From Pediatric Dilated Cardiomyopathy Patients Causes Dysregulation of Cardiolipin Biosynthesis and Mitochondrial Function Julie Pires Da Silva, Anastacia M. Garcia, Carissa A. Miyano, Genevieve C. Sparagna, Raleigh Jonscher, Hanan Elajaili, and Carmen C. Sucharov. University of Colorado Anschutz Medical Campus, Aurora, CO Dilated Cardiomyopathy (DCM) Hypothesis - Dilated cardiomyopathy (DCM) is defined as a disorder characterized by Using a novel in vitro model of DCM-related cardiomyocyte remodeling that dilation and impaired contraction of the left ventricle or both ventricles. reproduces the molecular characteristics of pediatric DCM, we hypothesized that the alteration of mitochondrial function in NRVM treated - DCM is the most common form of cardiomyopathy and cause of heart with DCM pediatric sera is associate with changes in cardiolipin content and failure in children older than 1 year of age with an annual incidence of 0.57 mitochondrial β-oxidation pathway. per 100,000 children. - The causes of heart failure (HF) in children differ substantially from those Results found in the adult population and children do not respond well to adult HF therapies. Cardiolipin (CL) - Cardiolipin is a mitochondrial dimeric phospholipid normally located in the inner mitochondrial membrane Figure 5. DCM serum induces significant changes in metabolite levels involved in fatty acid oxidation pathway in NRVMs. A. Heatmap of 41 - CL represent 12-15% of phospholipid mass in heart. In the metabolites differentially expressed in NF and DCM serum-treated NRVMs. n = 4 NF, n= 4 DCM samples, p<0.05. B. Pathway enrichment map analysis heart, 70-80% is (18:2)4CL. of differential metabolites between NF and DCM groups using Figure 3. -

Altered Traffic of Cardiolipin During Apoptosis: Exposure on the Cell Surface As a Trigger for (Antiphospholipid Antibodies)
Hindawi Publishing Corporation Journal of Immunology Research Volume 2015, Article ID 847985, 9 pages http://dx.doi.org/10.1155/2015/847985 Review Article Altered Traffic of Cardiolipin during Apoptosis: Exposure on the Cell Surface as a Trigger for (Antiphospholipid Antibodies) Valeria Manganelli,1 Antonella Capozzi,1 Serena Recalchi,1 Michele Signore,2 Vincenzo Mattei,1,3 Tina Garofalo,1 Roberta Misasi,1 Mauro Degli Esposti,4 and Maurizio Sorice1 1 Department of Experimental Medicine, Sapienza University of Rome, Viale Regina Elena 324, 00161 Rome, Italy 2Department of Hematology, Oncology and Molecular Medicine, National Institute of Health, Viale Regina Elena 299, 00161 Rome, Italy 3Laboratory of Experimental Medicine and Environmental Pathology, Sabina Universitas, Via dell’Elettronica, 02100 Rieti, Italy 4Italian Institute of Technology, Via Morego 30, 16136 Genoa, Italy Correspondence should be addressed to Maurizio Sorice; [email protected] Received 27 July 2015; Accepted 6 September 2015 Academic Editor: Douglas C. Hooper Copyright © 2015 Valeria Manganelli et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Apoptosis has been reported to induce changes in the remodelling of membrane lipids; after death receptor engagement, specific changes of lipid composition occur not only at the plasma membrane, but also in intracellular membranes. This paper focuses on one important aspect of apoptotic changes in cellular lipids, namely, the redistribution of the mitochondria-specific phospholipid, cardiolipin (CL). CL predominantly resides in the inner mitochondrial membrane, even if the rapid remodelling of its acyl chains and the subsequent degradation occur in other membrane organelles. -

Cops5 Safeguards Genomic Stability of Embryonic Stem Cells Through Regulating Cellular Metabolism and DNA Repair
Cops5 safeguards genomic stability of embryonic stem cells through regulating cellular metabolism and DNA repair Peng Lia, Lulu Gaoa, Tongxi Cuia, Weiyu Zhanga, Zixin Zhaoa, and Lingyi Chena,1 aState Key Laboratory of Medicinal Chemical Biology, Collaborative Innovation Center of Tianjin for Medical Epigenetics, Collaborative Innovation Center for Biotherapy, Tianjin Key Laboratory of Protein Sciences, National Demonstration Center for Experimental Biology Education and College of Life Sciences, Nankai University, 300071 Tianjin, China Edited by Janet Rossant, Hospital for Sick Children, University of Toronto, Toronto, Canada, and approved December 24, 2019 (received for review August 29, 2019) The highly conserved COP9 signalosome (CSN), composed of 8 transiently expressed in about 5% of ESCs at a given time, subunits (Cops1 to Cops8), has been implicated in pluripotency promotes rapid telomere elongation by telomere recombination maintenance of human embryonic stem cells (ESCs). Yet, the mech- and regulates genomic stability (11). Induced by genotoxic stress, anism for the CSN to regulate pluripotency remains elusive. We Filia stimulates the PARP1 activity and relocates from centro- previously showed that Cops2, independent of the CSN, is essential somes to DNA damage sites and mitochondria to regulate DDR for the pluripotency maintenance of mouse ESCs. In this study, we and apoptosis (12). Sall4, a pluripotency transcription factor, set out to investigate how Cops5 and Cops8 regulate ESC differ- facilitates the ataxia telangiectasia-mutated activation in re- entiation and tried to establish Cops5 and Cops8 knockout (KO) sponse to DSBs (13). To minimize the ROS-induced genomic ESC lines by CRISPR/Cas9. To our surprise, no Cops5 KO ESC clones DNA damage, ESCs produce lower levels of mitochondrial ROS were identified out of 127 clones, while three Cops8 KO ESC lines and express higher levels of antioxidants than differentiated cells were established out of 70 clones. -
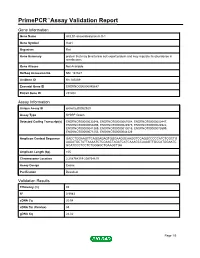
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name HCLS1-associated protein X-1 Gene Symbol Hax1 Organism Rat Gene Summary protein that may bind to bile salt export protein and may regulate its abundance in membranes Gene Aliases Not Available RefSeq Accession No. NM_181627 UniGene ID Rn.185269 Ensembl Gene ID ENSRNOG00000045647 Entrez Gene ID 291202 Assay Information Unique Assay ID qRnoCED0052920 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000030348, ENSRNOT00000067004, ENSRNOT00000000447, ENSRNOT00000059496, ENSRNOT00000024975, ENSRNOT00000024922, ENSRNOT00000041389, ENSRNOT00000010015, ENSRNOT00000073599, ENSRNOT00000071253, ENSRNOT00000044326 Amplicon Context Sequence GACCTGGAAGTTCAGGAGAGTGGGAAGGCAAGGTCCAGGCCCCCATCTCGCTG AAGATGCTATTAAAATCTCGAACTAGATCATCAAAGCCAAAGTTGCCATGGAATC GCATCCCTCCTCTGGGGCTGAAGCTGA Amplicon Length (bp) 105 Chromosome Location 2:208764319-208764619 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 92 R2 0.9983 cDNA Cq 20.54 cDNA Tm (Celsius) 83 gDNA Cq 24.02 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report Hax1, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Role of Calcium-Independent Phospholipase A2 in the Pathogenesis of Barth Syndrome
Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome Ashim Malhotraa, Irit Edelman-Novemskyb, Yang Xua, Heide Pleskenb, Jinping Mab, Michael Schlamea,b, and Mindong Renb,1 Departments of bCell Biology and aAnesthesiology, New York University Langone Medical Center, New York, NY 10016 Edited by David D. Sabatini, New York University School of Medicine, New York, NY, and approved December 23, 2008 (received for review November 6, 2008) Quantitative and qualitative alterations of mitochondrial cardio- is required for maintaining not only the normal CL fatty acyl lipin have been implicated in the pathogenesis of Barth syndrome, composition, but also normal CL levels. an X-linked cardioskeletal myopathy caused by a deficiency in Although it has been established that tafazzin deficiency tafazzin, an enzyme in the cardiolipin remodeling pathway. We causes both Barth syndrome and a derangement of CL metab- have generated and previously reported a tafazzin-deficient Dro- olism, evidence that it is, in fact, the CL deficiency that con- sophila model of Barth syndrome that is characterized by low tributes to Barth syndrome has been circumstantial (15). To cardiolipin concentration, abnormal cardiolipin fatty acyl compo- elucidate the pathogenic mechanism of Barth syndrome and to sition, abnormal mitochondria, and poor motor function. Here, we identify potential targets for therapeutic intervention, we have first show that tafazzin deficiency in Drosophila disrupts the final created a Drosophila model of Barth syndrome (7) by knocking stage of spermatogenesis, spermatid individualization, and causes out the tafazzin gene and have asked whether the resulting male sterility. This phenotype can be genetically suppressed by phenotypic changes can be suppressed by partially restoring CL inactivation of the gene encoding a calcium-independent phos- homeostasis without correcting the tafazzin defect. -

Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/Ipla2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters
antioxidants Review Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/iPLA2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters Martin Jab ˚urek 1,* , Pavla Pr ˚uchová 1, Blanka Holendová 1 , Alexander Galkin 2 and Petr Ježek 1 1 Department of Mitochondrial Physiology, Institute of Physiology of the Czech Academy of Sciences, Vídeˇnská 1084, 14220 Prague, Czech Republic; [email protected] (P.P.); [email protected] (B.H.); [email protected] (P.J.) 2 Department of Pediatrics, Division of Neonatology, Columbia University William Black Building, New York, NY 10032, USA; [email protected] * Correspondence: [email protected]; Tel.: +420-296442789 Abstract: Patatin-like phospholipase domain-containing protein PNPLA8, also termed Ca2+-independent phospholipase A2γ (iPLA2γ), is addressed to the mitochondrial matrix (or peroxisomes), where it may manifest its unique activity to cleave phospholipid side-chains from both sn-1 and sn-2 posi- tions, consequently releasing either saturated or unsaturated fatty acids (FAs), including oxidized FAs. Moreover, iPLA2γ is directly stimulated by H2O2 and, hence, is activated by redox signaling or oxidative stress. This redox activation permits the antioxidant synergy with mitochondrial un- coupling proteins (UCPs) or other SLC25 mitochondrial carrier family members by FA-mediated Citation: Jab ˚urek, M.; Pr ˚uchová,P.; protonophoretic activity, termed mild uncoupling, that leads to diminishing of mitochondrial su- Holendová, B.; Galkin, A.; Ježek, P. peroxide formation. This mechanism allows for the maintenance of the steady-state redox status of Antioxidant Synergy of the cell. Besides the antioxidant role, we review the relations of iPLA2γ to lipid peroxidation since Mitochondrial Phospholipase iPLA2γ is alternatively activated by cardiolipin hydroperoxides and hypothetically by structural γ PNPLA8/iPLA2 with Fatty alterations of lipid bilayer due to lipid peroxidation. -

Downloaded As a Non-Linear Data Structure Containing Ordered Pairs of 68 Proteins and All the Other Proteins with Which They Interact
bioRxiv preprint doi: https://doi.org/10.1101/854695; this version posted November 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Network potential identifies therapeutic miRNA cocktails in Ewings Sarcoma Davis T. Weaver1, 2, *, Kathleen I. Pishas3,*, Drew Williamson4,*, Jessica Scarborough1, 2, Stephen L. Lessnick3, Andrew Dhawan2, 5, †, and Jacob G. Scott1, 2, 6, † 1Case Western Reserve University School of Medicine, Cleveland, OH, 44106, USA 2Translational Hematology Oncology Research, Cleveland Clinic, Cleveland OH, 44106, USA 3Nationwide Children’s Hospital, Columbus, Ohio, 43205 4Department of Pathology, Massachusetts General Hospital, Boston, MA, 02144 5Division of Neurology, Cleveland Clinic, Cleveland Ohio, 44195 6Department of Physics, Case Western Reserve University, Cleveland, OH, 44106, USA *contributed equally †[email protected], [email protected] ABSTRACT Introduction: Micro-RNA (miRNA)-based therapies are an emerging class of cancer therapies with many potential applications in the field owing to their ability to repress multiple, predictable targets and cause widespread changes in a cell signaling network. New miRNA-based oligonucleotide drugs have have shown significant promise for the treatment of cancer in pre-clinical studies. Because of the broad effects miRNAs can have on different cells and tissues, a network science-based approach is well-equipped to evaluate and identify miRNA candidates and combinations of candidates for the repression of key oncogenic targets. Methods: In this work, we present a novel network science-based approach for identification of potential miRNA therapies, using Ewings Sarcoma as a model system. -

Duke University Dissertation Template
Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 i v ABSTRACT Gene-Environment Interactions in Cardiovascular Disease by Cavin Keith Ward-Caviness Graduate Program in Computational Biology and Bioinformatics Duke University Date:_______________________ Approved: ___________________________ Elizabeth R. Hauser, Supervisor ___________________________ William E. Kraus ___________________________ Sayan Mukherjee ___________________________ H. Frederik Nijhout An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Program in Computational Biology and Bioinformatics in the Graduate School of Duke University 2014 Copyright by Cavin Keith Ward-Caviness 2014 Abstract In this manuscript I seek to demonstrate the importance of gene-environment interactions in cardiovascular disease. This manuscript contains five studies each of which contributes to our understanding of the joint impact of genetic variation