Identification of the Fungal Catabolic D-Galacturonate Pathway
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sulfite Dehydrogenases in Organotrophic Bacteria : Enzymes
Sulfite dehydrogenases in organotrophic bacteria: enzymes, genes and regulation. Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften (Dr. rer. nat.) an der Universität Konstanz Fachbereich Biologie vorgelegt von Sabine Lehmann Tag der mündlichen Prüfung: 10. April 2013 1. Referent: Prof. Dr. Bernhard Schink 2. Referent: Prof. Dr. Andrew W. B. Johnston So eine Arbeit wird eigentlich nie fertig, man muss sie für fertig erklären, wenn man nach Zeit und Umständen das möglichste getan hat. (Johann Wolfgang von Goethe, Italienische Reise, 1787) DANKSAGUNG An dieser Stelle möchte ich mich herzlich bei folgenden Personen bedanken: . Prof. Dr. Alasdair M. Cook (Universität Konstanz, Deutschland), der mir dieses Thema und seine Laboratorien zur Verfügung stellte, . Prof. Dr. Bernhard Schink (Universität Konstanz, Deutschland), für seine spontane und engagierte Übernahme der Betreuung, . Prof. Dr. Andrew W. B. Johnston (University of East Anglia, UK), für seine herzliche und bereitwillige Aufnahme in seiner Arbeitsgruppe, seiner engagierten Unter- stützung, sowie für die Übernahme des Koreferates, . Prof. Dr. Frithjof C. Küpper (University of Aberdeen, UK), für seine große Hilfsbereitschaft bei der vorliegenden Arbeit und geplanter Manuskripte, als auch für die mentale Unterstützung während der letzten Jahre! Desweiteren möchte ich herzlichst Dr. David Schleheck für die Übernahme des Koreferates der mündlichen Prüfung sowie Prof. Dr. Alexander Bürkle, für die Übernahme des Prüfungsvorsitzes sowie für seine vielen hilfreichen Ratschläge danken! Ein herzliches Dankeschön geht an alle beteiligten Arbeitsgruppen der Universität Konstanz, der UEA und des SAMS, ganz besonders möchte ich dabei folgenden Personen danken: . Dr. David Schleheck und Karin Denger, für die kritische Durchsicht dieser Arbeit, der durch und durch sehr engagierten Hilfsbereitschaft bei Problemen, den zahlreichen wissenschaftlichen Diskussionen und für die aufbauenden Worte, . -

Genomewide and Enzymatic Analysis Reveals Efficient D-Galacturonic
RESEARCH ARTICLE Molecular Biology and Physiology Genomewide and Enzymatic Analysis Reveals Efficient D-Galacturonic Acid Metabolism in the Basidiomycete Yeast Rhodosporidium toruloides Ryan J. Protzko,a,b Christina A. Hach,c Samuel T. Coradetti,b,d Magdalena A. Hackhofer,c Sonja Magosch,c Nils Thieme,c Gina M. Geiselman,b,d Adam P. Arkin,b,d,f Jeffrey M. Skerker,b,d,f John E. Dueber,b,e,f J. Philipp Benzc aDepartment of Molecular and Cell Biology, University of California, Berkeley, California, USA bEnergy Biosciences Institute, Berkeley, California, USA cHolzforschung München, TUM School of Life Sciences Weihenstephan, Technische Universität München, Freising, Germany dEnvironmental Genomics and Systems Biology Division, Lawrence Berkeley National Laboratory, Berkeley, California, USA eBiological Systems & Engineering Division, Lawrence Berkeley National Laboratory, Berkeley, California, USA fDepartment of Bioengineering, University of California, Berkeley, California, USA ABSTRACT Biorefining of renewable feedstocks is one of the most promising routes to replace fossil-based products. Since many common fermentation hosts, such as Saccharomyces cerevisiae, are naturally unable to convert many component plant cell wall polysaccharides, the identification of organisms with broad catabolism capabilities represents an opportunity to expand the range of substrates used in fer- mentation biorefinery approaches. The red basidiomycete yeast Rhodosporidium to- ruloides is a promising and robust host for lipid- and terpene-derived chemicals. Pre- vious studies demonstrated assimilation of a range of substrates, from C5/C6 sugars to aromatic molecules similar to lignin monomers. In the current study, we analyzed the potential of R. toruloides to assimilate D-galacturonic acid, a major sugar in many pectin-rich agricultural waste streams, including sugar beet pulp and citrus peels. -

Xylooligosaccharides Production, Quantification, and Characterization
19 Xylooligosaccharides Production, Quantification, and Characterization in Context of Lignocellulosic Biomass Pretreatment Qing Qing1, Hongjia Li2,3,4,Ã, Rajeev Kumar2,4 and Charles E. Wyman2,3,4 1 Pharmaceutical Engineering & Life Science, Changzhou University, Changzhou, China 2 Center for Environmental Research and Technology, University of California, Riverside, USA 3 Department of Chemical and Environmental Engineering, University of California, Riverside, USA 4 BioEnergy Science Center, Oak Ridge, USA 19.1 Introduction 19.1.1 Definition of Oligosaccharides Oligosaccharides, also termed sugar oligomers, refer to short-chain polymers of monosaccharide units con- nected by a and/or b glycosidic bonds. In structure, oligosaccharides represent a class of carbohydrates between polysaccharides and monosaccharides, but the range of degree of polymerization (DP, chain length) spanned by oligosaccharides has not been consistently defined. For example, the Medical Subject Headings (MeSH) database of the US National Library of Medicine defines oligosaccharides as carbohy- drates consisting of 2–10 monosaccharide units; in other literature, sugar polymers with DPs of up to 30–40 have been included as oligosaccharides [1–3]. ÃPresent address: DuPont Industrial Biosciences, Palo Alto, USA Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, First Edition. Edited by Charles E. Wyman. Ó 2013 John Wiley & Sons, Ltd. Published 2013 by John Wiley & Sons, Ltd. 392 Aqueous Pretreatment of Plant Biomass for -
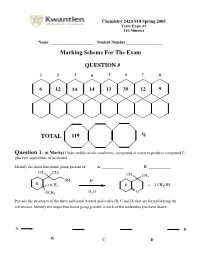
Marking Scheme for the Exam
Chemistry 2420 S10 Spring 2005 Term Exam #1 110 Minutes Name: ______________________ Student Number___________________ Marking Scheme For The Exam QUESTION # 1 2 3 4 5 6 7 8 6 12 14 14 13 39 12 9 TOTAL 119 % Question 1. (6 Marks) Under mildly acidic conditions, compound A reacts to produce compound E plus two equivalents of methanol. Identify the main functional group present in: A: ___________ E: ___________ CH3 CH3 CH 3 CH3 + NH2 H A OCH3 E + 2 CH3OH H O OCH3 2 N Provide the structures of the three additional neutral molecules (B, C and D) that are formed during the conversion. Identify the major functional group present in each of the molecules you have drawn. A E B C D Question 2. (12 Marks) Circle the structure that will best satisfy the given information. - the compound able to undergo an intramolecular cyclization to form a hemiacetal H H HO H HO OH O O O - the compound able to undergo an intramolecular cyclization to form an enamine H H H Me2N MeHN H2N O O O + - the produc t formed from treatment of 2-butanone with NaBD4 followed by a H3O work-up DO H HO D DO D HO H - the compound that would most likely be coloured O H O O - the compound capable of undergoing mutarotation O O O OH OCH3 OH CH3 H CH 3 - the compound that would not react with MeMgBr O O O OH OCH3 OH CH3 H CH3 - the compound bes t described as “anti-aromatic”: - the most stable carbocation + + + Cl Cl Cl OCH 3 H NO2 H H H Question 3. -

Assembly of an Active Enzyme by the Linkage of Two Protein Modules
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 1069–1073, February 1997 Biochemistry Assembly of an active enzyme by the linkage of two protein modules A. E. NIXON,M.S.WARREN, AND S. J. BENKOVIC* 152 Davey Laboratory, Department of Chemistry, Pennsylvania State University, University Park, PA 16802-6300 Contributed by S. J. Benkovic, December 9, 1996 ABSTRACT The feasibility of creating new enzyme activ- design enzymes with novel properties. Previous approaches to ities from enzymes of known function has precedence in view the design of proteins with novel activities have included of protein evolution based on the concepts of molecular catalytic antibodies (8, 9); introduction of metal ion binding recruitment and exon shuffling. The enzymes encoded by the sites, such as the one engineered into trypsin to allow either Escherichia coli genes purU and purN, N10-formyltetrahydro- control of the proteolytic activity (10) or to regulate specificity folate hydrolase and glycinamide ribonucleotide (GAR) trans- (11); creation of hybrid enzymes through exchange of subunits formylase, respectively, catalyze similiar yet distinct reactions. to create hybrid oligomers (12); replacement of structural N10-formyltetrahydrofolate hydrolase uses water to cleave elements such as the DNA binding domain of GCN4 with that N10-formyltetrahydrofolate into tetrahydrofolate and for- of CyEBP (13); mutation of multiple individual residues to mate, whereas GAR transformylase catalyses the transfer of change the cofactor specificity of glutathione reductase from formyl from N10-formyltetrahydrofolate to GAR to yield NADPH to NADH (14), modulation of the substrate speci- formyl-GAR and tetrahydrofolate. The two enzymes show ficity of aspartate aminotransferase (15); and changing the significant homology ('60%) in the carboxyl-terminal region specificity of subtilisin (16) and a-lytic protease (17) through which, from the GAR transformylase crystal structure and mutation of single functional groups. -

Resolution of Carbon Metabolism and Sulfur-Oxidation Pathways of Metallosphaera Cuprina Ar-4 Via Comparative Proteomics
JOURNAL OF PROTEOMICS 109 (2014) 276– 289 Available online at www.sciencedirect.com ScienceDirect www.elsevier.com/locate/jprot Resolution of carbon metabolism and sulfur-oxidation pathways of Metallosphaera cuprina Ar-4 via comparative proteomics Cheng-Ying Jianga, Li-Jun Liua, Xu Guoa, Xiao-Yan Youa, Shuang-Jiang Liua,c,⁎, Ansgar Poetschb,⁎⁎ aState Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, PR China bPlant Biochemistry, Ruhr University Bochum, Bochum, Germany cEnvrionmental Microbiology and Biotechnology Research Center, Institute of Microbiology, Chinese Academy of Sciences, Beijing, PR China ARTICLE INFO ABSTRACT Article history: Metallosphaera cuprina is able to grow either heterotrophically on organics or autotrophically Received 16 March 2014 on CO2 with reduced sulfur compounds as electron donor. These traits endowed the species Accepted 6 July 2014 desirable for application in biomining. In order to obtain a global overview of physiological Available online 14 July 2014 adaptations on the proteome level, proteomes of cytoplasmic and membrane fractions from cells grown autotrophically on CO2 plus sulfur or heterotrophically on yeast extract Keywords: were compared. 169 proteins were found to change their abundance depending on growth Quantitative proteomics condition. The proteins with increased abundance under autotrophic growth displayed Bioleaching candidate enzymes/proteins of M. cuprina for fixing CO2 through the previously identified Autotrophy 3-hydroxypropionate/4-hydroxybutyrate cycle and for oxidizing elemental sulfur as energy Heterotrophy source. The main enzymes/proteins involved in semi- and non-phosphorylating Entner– Industrial microbiology Doudoroff (ED) pathway and TCA cycle were less abundant under autotrophic growth. Also Extremophile some transporter proteins and proteins of amino acid metabolism changed their abundances, suggesting pivotal roles for growth under the respective conditions. -

REFERENCE ONLY UNIVERSITY of LONDON THESIS This Thesis
REFERENCE ONLY UNIVERSITY OF LONDON THESIS Degree Year Name of Author 0" * COPYRIGHT This is a thesis accepted for a Higher Degree of the University of London. It is an unpublished typescript and the copyright is held by the author. All persons consulting the thesis must read and abide by the Copyright Declaration below. COPYRIGHT DECLARATION I recognise that the copyright of the above-described thesis rests with the author and that no quotation from it or information derived from it may be published without the prior written consent of the author. LOANS Theses may not be lent to individuals, but the Senate House Library may lend a copy to approved libraries within the United Kingdom, for consultation solely on the premises of those libraries. Application should be made to: Inter-Library Loans, Senate House Library, Senate House, Malet Street, London WC1E 7HU. REPRODUCTION University of London theses may not be reproduced without explicit written permission from the Senate House Library. Enquiries should be addressed to the Theses Section of the Library. Regulations concerning reproduction vary according to the date of acceptance of the thesis and are listed below as guidelines. A. Before 1962. Permission granted only upon the prior written consent of the author. (The Senate House Library will provide addresses where possible). B. 1962- 1974. In many cases the author has agreed to permit copying upon completion of a Copyright Declaration. C. 1975 - 1988. Most theses may be copied upon completion of a Copyright Declaration. D. 1989 onwards. Most theses may be copied. This thesis comes within category D. -

Dissimilation of Cysteate Via 3-Sulfolactate Sulfo-Lyase and a Sulfate Exporter in Paracoccus Pantotrophus NKNCYSA
Microbiology (2005), 151, 737–747 DOI 10.1099/mic.0.27548-0 Dissimilation of cysteate via 3-sulfolactate sulfo-lyase and a sulfate exporter in Paracoccus pantotrophus NKNCYSA Ulrike Rein,1 Ronnie Gueta,1 Karin Denger,1 Ju¨rgen Ruff,1 Klaus Hollemeyer2 and Alasdair M. Cook1 Correspondence 1Department of Biology, The University, D-78457 Konstanz, Germany Alasdair Cook 2Institute of Biochemical Engineering, Saarland University, Box 50 11 50, D-66041 [email protected] Saarbru¨cken, Germany Paracoccus pantotrophus NKNCYSA utilizes (R)-cysteate (2-amino-3-sulfopropionate) as a sole source of carbon and energy for growth, with either nitrate or molecular oxygen as terminal electron acceptor, and the specific utilization rate of cysteate is about 2 mkat (kg protein)”1. The initial degradative reaction is catalysed by an (R)-cysteate : 2-oxoglutarate aminotransferase, which yields 3-sulfopyruvate. The latter was reduced to 3-sulfolactate by an NAD-linked sulfolactate dehydrogenase [3?3 mkat (kg protein)”1]. The inducible desulfonation reaction was not detected initially in cell extracts. However, a strongly induced protein with subunits of 8 kDa (a) and 42 kDa (b) was found and purified. The corresponding genes had similarities to those encoding altronate dehydratases, which often require iron for activity. The purified enzyme could then be shown to convert 3-sulfolactate to sulfite and pyruvate and it was termed sulfolactate sulfo-lyase (Suy). A high level of sulfite dehydrogenase was also induced during growth with cysteate, and the organism excreted sulfate. A putative regulator, OrfR, was encoded upstream of suyAB on the reverse strand. Downstream of suyAB was suyZ, which was cotranscribed with suyB. -

Part 1 in Our Series of Carbohydrate Lectures. in This Section, You Will Learn About Monosaccharide Structure
Welcome to Part 1 in our series of Carbohydrate lectures. In this section, you will learn about monosaccharide structure. The building blocks of larger carbohydrate polymers. 1 First, let’s review why learning about carbohydrates is important. Carbohydrates are used by biological systems as fuels and energy resources. Carbohydrates typically provide quick energy and are one of the primary energy storage forms in animals. Carbohydrates also provide the precursors to other major macromolecules within the body, including the deoxyribose and ribose required for nucleic acid biosynthesis. Carbohydrates can also provide structural support and cushioning/shock absorption, as well as cell‐cell communication, identification, and signaling. 2 Carbohydrates, as their name implies, are water hydrates of carbon, and they all have the same basic core formula (CH2O)n and are always found in the ratio of 1 carbon to 2 hydrogens to 1 oxygen (1:2:1) making them easy to identify from their molecular formula. 3 Carbohydrates can be divided into subcategories based on their complexity. The simplest carbohydrates are the monosaccharides which are the simple sugars required for the biosynthesis of all the other carbohydrate types. Disaccharides consist of two monosaccharides that have been joined together by a covalent bond called the glycosidic bond. Oligosaccharides are polymers that consist of a few monosaccharides covalently linked together, and Polysaccharides are large polymers that contain hundreds to thousands of monosaccharide units all joined together by glycosidic bonds. The remainder of this lecture will focus on monosaccharides 4 Monosaccharides all have alcohol functional groups associated with them. In addition they also have one additional functional group, either an aldehyde or a ketone. -
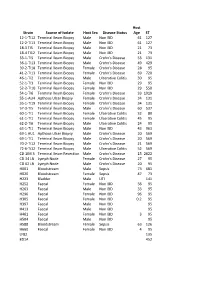
Strain Source of Isolate Host Sex Disease Status Host Age ST
Host Strain Source of Isolate Host Sex Disease Status Age ST 12-1-TI12 Terminal Ileum Biopsy Male Non IBD 61 127 12-2-TI13 Terminal Ileum Biopsy Male Non IBD 61 127 18-3 TI5 Terminal Ileum Biopsy Male Non IBD 21 73 18-4 TI12 Terminal Ileum Biopsy Male Non IBD 21 73 33-1-TI5 Terminal Ileum Biopsy Male Crohn's Disease 53 131 36-1-TI13 Terminal Ileum Biopsy Male Crohn's Disease 49 429 39-2-TI18 Terminal Ileum Biopsy Female Crohn's Disease 28 95 41-2-TI13 Terminal Ileum Biopsy Female Crohn's Disease 69 720 46-1-TI2 Terminal Ileum Biopsy Male Ulcerative Colitis 30 95 52-1-TI3 Terminal Ileum Biopsy Female Non IBD 29 95 52-2-TI10 Terminal Ileum Biopsy Female Non IBD 29 550 54-1-TI6 Terminal Ileum Biopsy Female Crohn's Disease 33 1919 55-1-AU4 Apthous Ulcer Biopsy Female Crohn's Disease 34 131 55-1-TI19 Terminal Ileum Biopsy Female Crohn's Disease 34 131 57-3-TI5 Terminal Ileum Biopsy Male Crohn's Disease 60 537 60-1-TI1 Terminal Ileum Biopsy Female Ulcerative Colitis 32 80 61-1-TI1 Terminal Ileum Biopsy Female Ulcerative Colitis 45 95 62-2-TI6 Terminal Ileum Biopsy Male Ulcerative Colitis 24 95 63-1-TI1 Terminal Ileum Biopsy Male Non IBD 43 963 69-1 AU1 Apthous Ulcer Biopsy Male Crohn's Disease 20 569 69-1-TI1 Terminal Ileum Biopsy Male Crohn's Disease 20 569 70-2-TI12 Terminal Ileum Biopsy Male Crohn's Disease 21 569 72-6-Ti12 Terminal Ileum Biopsy Male Ulcerative Colitis 52 569 CD 1IM 3 Terminal Ileum Resection Male Crohn's Disease 15 2622 CD 34 LN Lymph Node Female Crohn's Disease 27 95 CD 62 LN Lymph Node Male Crohn's Disease 20 95 H001 -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Lipids, Carbohydrates, Nucleobases & DNA
Chemistry 2050 “Introduction to Organic Chemistry” Fall Semester 2011 Dr. Rainer Glaser Examination #5: The Final “Lipids, Carbohydrates, Nucleobases & DNA.” Monday, December 12, 2011, 10 am – 12 pm. Name: Answer Key Question 1. Lipids and Detergents. 20 Question 2. Glyceraldehyde and Carbohydrates. 20 Question 3. Nucleobases of DNA and RNA. 20 Question 4. Ribose and Deoxyribose and Their Phosphate Esters. 20 Question 5. Single and Double Strands of DNA. 20 Total 100 — 1 — Question 1. Lipids and Detergents. (20 points) (a) Draw the line segment drawing of glyceryl distearoleate. This fat is the _TRI-ester formed between one molecule of the triol __GLYCEROL_, two molecules of the fatty acid stearic acid, H3C−(CH2)16−COOH, and one molecule of the unsaturated fatty acid oleic acid, H3C−(CH2)7−CH=CH−(CH2)7−COOH. In your drawing, attach the oleic acid to one of the primary alcohols of the triol. Clearly indicate whether the alkene in oleic acid is cis or trans. To convert this fat into soap, one would cook the fat with aqueous NaOH solution, a process that is called __SAPONIFICATION__. (12 points) (b) A simple micelle is shown schematically on the right. Each circle signifies a _________ (polar, nonpolar) head-group, and each wiggly line signifies a ________ (polar, nonpolar) alkyl chain. In the case of “normal” soap, the head- group is a ___CARBOXYLATE__ anion. Indicate where we would find the associated cations (i.e., sodium cations; draw a few in the scheme). Water is on the __________ (inside, outside) of this micelle and fats will accumulate on the _________ (inside, outside) of this micelle.