Microglia and C9orf72 in Neuroinflammation and ALS and Frontotemporal Dementia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inside Your Brain You and Your Brain
Inside your brain You and your brain Many simple and complex psychological functions are mediated by multiple brain regions and, at the same time, a single brain area may control many psychological functions. CC BY Illustration by Bret Syfert 1. Cortex: The thin, folded structure on the outside surface of the brain. 2. Cerebral hemispheres: The two halves of the brain, each of which controls and receives information from the opposite side of the body. 3. Pituitary gland: The ‘master gland’ of the body, which releases hormones that control growth, blood pressure, the stress response and the function of the sex organs. 4. Substantia nigra: The ‘black substance’ contains cells that produce the neurotransmitter dopamine and the pigment melatonin, giving it a black appearance. 5. Hypothalamus: The interface between the brain and pituitary gland. It controls the production and release of hormones. 6. Spinal cord: A large bundle of millions of nerve fibres and neuronal cells, which carries information back and forth between the brain and the body. 7. Medulla oblongata: Controls vital involuntary functions such as breathing and heart rate. 8. Cerebellum: The ‘little brain’ that controls balance and coordinates movements. It’s normally required for learning motor skills, such as riding a bike, and is involved in thought processes. 9. Cranial nerve nuclei: Clusters of neurons in the brain stem. Their axons form the cranial nerves. Your brain underpins who you are. It stores your knowledge and memories, gives you the capacity for thought and emotion, and enables you to control your body. The brain is just one part of the nervous system. -

Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems
cells Review Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems Naoki Abe 1, Tasuku Nishihara 1,*, Toshihiro Yorozuya 1 and Junya Tanaka 2 1 Department of Anesthesia and Perioperative Medicine, Ehime University Graduate School of Medicine, Toon, Ehime 791-0295, Japan; [email protected] (N.A.); [email protected] (T.Y.) 2 Department of Molecular and cellular Physiology, Ehime University Graduate School of Medicine, Toon, Ehime 791-0295, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-89-960-5383; Fax: +81-89-960-5386 Received: 4 August 2020; Accepted: 17 September 2020; Published: 21 September 2020 Abstract: Microglia, the immunocompetent cells in the central nervous system (CNS), have long been studied as pathologically deteriorating players in various CNS diseases. However, microglia exert ameliorating neuroprotective effects, which prompted us to reconsider their roles in CNS and peripheral nervous system (PNS) pathophysiology. Moreover, recent findings showed that microglia play critical roles even in the healthy CNS. The microglial functions that normally contribute to the maintenance of homeostasis in the CNS are modified by other cells, such as astrocytes and infiltrated myeloid cells; thus, the microglial actions on neurons are extremely complex. For a deeper understanding of the pathophysiology of various diseases, including those of the PNS, it is important to understand microglial functioning. In this review, we discuss both the favorable and unfavorable roles of microglia in neuronal survival in various CNS and PNS disorders. We also discuss the roles of blood-borne macrophages in the pathogenesis of CNS and PNS injuries because they cooperatively modify the pathological processes of resident microglia. -

Gamma Motor Neurons Survive and Exacerbate Alpha Motor Neuron Degeneration In
Gamma motor neurons survive and exacerbate alpha PNAS PLUS motor neuron degeneration in ALS Melanie Lalancette-Heberta,b, Aarti Sharmaa,b, Alexander K. Lyashchenkoa,b, and Neil A. Shneidera,b,1 aCenter for Motor Neuron Biology and Disease, Columbia University, New York, NY 10032; and bDepartment of Neurology, Columbia University, New York, NY 10032 Edited by Rob Brownstone, University College London, London, United Kingdom, and accepted by Editorial Board Member Fred H. Gage October 27, 2016 (received for review April 4, 2016) The molecular and cellular basis of selective motor neuron (MN) the muscle spindle and control the sensitivity of spindle afferent vulnerability in amyotrophic lateral sclerosis (ALS) is not known. In discharge (15); beta (β) skeletofusimotor neurons innervate both genetically distinct mouse models of familial ALS expressing intra- and extrafusal muscle (16). In addition to morphological mutant superoxide dismutase-1 (SOD1), TAR DNA-binding protein differences, distinct muscle targets, and the absence of primary 43 (TDP-43), and fused in sarcoma (FUS), we demonstrate selective afferent (IA)inputsonγ-MNs, these functional MN subtypes also degeneration of alpha MNs (α-MNs) and complete sparing of differ in their trophic requirements, and γ-MNs express high levels gamma MNs (γ-MNs), which selectively innervate muscle spindles. of the glial cell line-derived neurotropic factor (GDNF) receptor Resistant γ-MNs are distinct from vulnerable α-MNs in that they Gfrα1 (17). γ-MNs are also molecularly distinguished by the ex- lack synaptic contacts from primary afferent (IA) fibers. Elimination pression of other selective markers including the transcription α of these synapses protects -MNs in the SOD1 mutant, implicating factor Err3 (18), Wnt7A (19), the serotonin receptor 1d (5-ht1d) this excitatory input in MN degeneration. -

ARIA Is Concentrated in Nerve Terminals at Neuromuscular Junctions and at Other Synapses
The Journal of Neuroscience, September 1995, 15(g): 6124-6136 ARIA Is Concentrated in Nerve Terminals at Neuromuscular Junctions and at Other Synapses Alfred W. Sandrock, Jr.,i,2 Andrew D. J. Goodearl,’ Qin-Wei Yin,’ David Chang,3 and Gerald D. Fischbachl ‘Department of Neurobiology, Harvard Medical School, Boston, Massachusetts 02115, “Department of Neurology, Massachusetts General Hosoital, Boston, Massachusetts 02114, and 3Amgen, Inc., Thousand Oaks, California 91320 Skeletal muscle ACh receptors (AChRs) accumulate at neu- natal maturation of AChRs at the neuromuscular junction, which romuscular junctions (nmjs) at least partly because of the results in the replacement of y-subunit-containing receptors with selective induction of AChR subunit genes in subsynaptic those containing E- subunits (Brenner and Sakmann, 1978; Bren- myotube nuclei by the motor nerve terminal. Additionally, ner et al., 1990; Martinou and Merlie, 1991). ARIA (for ACh mammalian AChRs undergo a postnatal change in subunit receptor inducing activity), which was purified from chicken composition from embryonic (cw.&S) to adult (@eS) brains based on its ability to stimulate the synthesis of AChRs forms, a switch that also depends on innervation. ARIA, a in skeletal muscle (Falls et al., 1993), may mediate the ability protein purified from chicken brains based on its ability to of the motor neuron to orchestrate these developmental events. induce AChR synthesis in primary chick muscle cells, is a ARIA mRNA is concentrated in chick and rat motor neurons strong candidate for being the molecule responsible for (Falls et al., 1993; Corfas et al., 1995), and, in both species, is these early developmental events. -

Perineurial Glia Require Notch Signaling During Motor Nerve Development but Not Regeneration
The Journal of Neuroscience, March 6, 2013 • 33(10):4241–4252 • 4241 Development/Plasticity/Repair Perineurial Glia Require Notch Signaling during Motor Nerve Development but Not Regeneration Laura A. Binari, Gwendolyn M. Lewis, and Sarah Kucenas Department of Biology, University of Virginia, Charlottesville, Virginia 22904 Motor nerves play the critical role of shunting information out of the CNS to targets in the periphery. Their formation requires the coordinated development of distinct cellular components, including motor axons and the Schwann cells and perineurial glia that en- sheath them. During nervous system assembly, these glial cells must migrate long distances and terminally differentiate, ensuring the efficient propagation of action potentials. Although we know quite a bit about the mechanisms that control Schwann cell development during this process, nothing is known about the mechanisms that mediate the migration and differentiation of perineurial glia. Using in vivo imaging in zebrafish, we demonstrate that Notch signaling is required for both perineurial migration and differentiation during nerve formation, but not regeneration. Interestingly, loss of Notch signaling in perineurial cells also causes a failure of Schwann cell differentiation, demonstrating that Schwann cells require perineurial glia for aspects of their own development. These studies describe a novel mechanism that mediates multiple aspects of perineurial development and reveal the critical importance of perineurial glia for Schwann cell maturation and nerve formation. Introduction independent. However, to date, nothing is known about these A fundamental goal in neuroscience is to understand what con- nonaxonal mechanisms. trols cell migration. Unlike other organ systems where cells are In zebrafish, perineurial glia originate from precursors in restricted to a discrete space, peripheral glia must migrate long the floor plate (p3 domain) of the spinal cord, migrate out of distances from their origin to their target. -

How Is the Brain Organized?
p CHAPTER 2 How Is the Brain Organized? An Overview of Brain Structure The Functional Organization Brain Terminology of the Brain The Brain’s Surface Features Principle 1: The Sequence of Brain Processing The Brain’s Internal Features Is “In Integrate Out” Microscopic Inspection: Cells and Fibers Principle 2: Sensory and Motor Divisions Exist Focus on Disorders: Meningitis and Throughout the Nervous System Encephalitis Principle 3: The Brain’s Circuits Are Crossed Focus on Disorders: Stroke Principle 4: The Brain Is Both Symmetrical and Asymmetrical Principle 5: The Nervous System Works A Closer Look at Neuroanatomy Through Excitation and Inhibition The Cranial Nervous System Principle 6: The Central Nervous System Has The Spinal Nervous System Multiple Levels of Function The Internal Nervous System Principle 7: Brain Systems Are Organized Both Focus on Disorders: Magendie, Bell, and Bell’s Hierarchically and in Parallel Palsy Principle 8: Functions in the Brain Are Both Localized and Distributed A. Klehr / Stone Images Micrograph: Carolina Biological Supply Co. / Phototake 36 I p hen buying a new car, people first inspect the In many ways, examining a brain for the first time is outside carefully, admiring the flawless finish similar to looking under the hood of a car. We have a vague W and perhaps even kicking the tires. Then they sense of what the brain does but no sense of how the parts open the hood and examine the engine, the part of the car that we see accomplish these tasks. We may not even be responsible for most of its behavior—and misbehavior. able to identify many of the parts. -

Astrocyte Manipulation Alters Mouse Behavior Keebum Park1 Andsungjoonglee2
Park and Lee Experimental & Molecular Medicine (2020) 52:1028–1038 https://doi.org/10.1038/s12276-020-0468-z Experimental & Molecular Medicine REVIEW ARTICLE Open Access Deciphering the star codings: astrocyte manipulation alters mouse behavior Keebum Park1 andSungJoongLee2 Abstract Astrocytes occupy a vast area within the central nervous system (CNS). Despite their abundance, the functional role of astrocytes in vivo has only begun to be uncovered. Astrocytes were typically thought to be involved in pathophysiological states. However, recent studies have shown that astrocytes are actively involved in cell signaling in normal physiological states; manipulating various aspects of astrocytic cell signaling in vivo has revealed that astrocytes are key players in controlling healthy behavior in the absence of pathophysiology. Unfortunately, the study of astrocyte function is often limited by the number of approaches available due to our lack of understanding of cell physiology. This review summarizes recent studies in which altered astrocyte signaling capacity resulted in dramatic changes in behavior. We not only discuss the methodologies available to manipulate astrocytes but also provide insights into the behavioral roles of astrocytes in the CNS. Introduction physiological activities. Recent studies have also recog- Astrocytes, or “star-shaped” cells, which are as abundant nized the importance of the functional and regulatory 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; in our CNS as neurons, densely and homogeneously roles that astrocytes play under normal physiological populate the brain, spinal cord, and retina1,2. Although conditions. The final outputs that produce one’s behavior astrocytes are abundantly distributed across the entire are generally accepted to be neuronal; however, astrocytes brain, they have traditionally been regarded as “support often critically modulate this final output. -
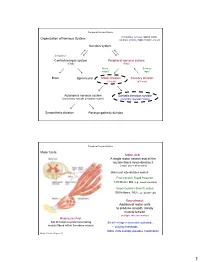
Nervous System Central Nervous System Peripheral Nervous System
Peripheral Nervous System Involuntary reflexes (spinal cord); Organization of Nervous System: voluntary actions (higher brain centers) Nervous system Integration Central nervous system Peripheral nervous system (CNS) (PNS) Motor Sensory output input Brain Spinal cord Motor division Sensory division (efferent) (afferent) Autonomic nervous system Somatic nervous system (involuntary; smooth & cardiac muscle) (voluntary; skeletal muscle) Sympathetic division Parasympathetic division Peripheral Nervous System Motor Units: Motor Unit: A single motor neuron and all the muscle fibers innervated by it (motor unit = all-or-none) Motor unit size dictates control: Fine Control / Rapid Reaction: 1-10 fibers / MU (e.g., ocular muscles) Gross Control / Slow Reaction: 1000’s fibers / MU (e.g., quadriceps) Recruitment: Addition of motor units to produce smooth, steady muscle tension (multiple fiber summation) Motoneuron Pool: Set of motor neurons innervating Small large motor units activated… muscle fibers within the same muscle • Varying thresholds Motor units overlap; provides coordination Marieb & Hoehn – Figure 9.13 1 Peripheral Nervous System Types of Motor Neurons: 1) Alpha () motor neurons: • Give rise to large Type A alpha (A) motor nerve fibers (~ 14 µm diameter) • Innervate extrafusal skeletal muscle fibers (generate force) 2) Gamma () motor neurons: • Give rise to small Type A gamma (Aγ) motor nerve fibers (~ 5 µm diameter) • Innervate intrafusal muscle fibers (small, specialized fibers – muscle spindle) What is the length of the muscle? Proper -

The Nervous System Unit One: Source
UNIT ONE THE NERVOUS SYSTEM Unit One: The Nervous System UNC-CH Brain Explorers May be reproduced for non-profit educational use only. Please credit source. 3 UNIT ONE THE NERVOUS SYSTEM Unit One: The Nervous System UNC-CH Brain Explorers May be reproduced for non-profit educational use only. Please credit source. 4 THE NERVOUS SYSTEM UNIFYING SUMMARY KEY POINTS CONCEPTS FULL BODY TRACING • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • • Models help us understand and In the Full Body Tracing students • All our thooughts, movements, explain the world.* work together as a class to create sensations and emotions are • Systems are made of parts which a model of the nervous system. controlled by the nervous system. connect to create the whole.* The outline of a student’s body is • We have a Central Nervous • The brain receives informational traced on a large piece of paper. System (brain and spinal cord) signals from all parts of the body. A cardboard cut out of the brain and Peripheral Nervous System The brain sends signals to all parts and spinal cord are placed in the (nerves extending from the spinal of the body to influence what they outline to represent the central cord to limbs, trunk, face, organs do.* nervous system. Two colors of and throughout.) • Humans have systems for yarn, representing the motor and • Sensory nerves communicate digestion, circulation, movement sensory nerves, are used to create information from the body to the and coordination. These systems motor and sensory pathways of brain and motor nerves, from the interact with one another.* the peripheral nervous system. brain to the body. -

NERVOUS SYSTEM ANATOMY M1 - Gross and Developmental Anatomy Dr
INTRODUCTION to NERVOUS SYSTEM ANATOMY M1 - Gross and Developmental Anatomy Dr. Milton M. Sholley Professor of Anatomy and Neurobiology and Dr. Michael H. Peters Professor of Chemical and Life Science Engineering Lecture/Syllabus Outline I. Major Divisions of the Nervous System II. Cranial Nerves (structure) III. Spinal Nerves (structure) IV. Autonomic Nervous System (functional) V. Functional Components of Spinal Nerves VI. Dermatomes 2 Important Definitions I. Major divisions of the nervous system include: A. Central nervous system (CNS)-an anatomical (i.e. structural) classification 1. Brain 2. Spinal cord B. Peripheral nervous system (PNS)-an anatomical (i.e. structural) classification 1. Cranial nerves-arise from the brain 2. Spinal nerves-arise from the spinal cord C. Somatic nervous system (SNS)-a functional classification. Parts of this system begin in the CNS and enter the PNS to supply the body wall (“soma”). D. Autonomic nervous system (ANS)-a functional classification. Parts of this system begin in the CNS and enter the PNS to supply the viscera. 1. Parasympathetic division 2. Sympathetic division Cranial Nerves (all twelve pairs shown) II. Cranial nerves A. Twelve pairs of cranial nerves arise from various parts of the brain and have both I numbers designated by Roman numerals and names. I IIII 1. Most of the cranial nerves will not be IV studied until you begin the Head and Neck V portion of the course, when they will be studied in detail. VI VII B. Cranial nerve XI, the spinal accessory VII nerve, has been introduced this week in regard I IX to the trapezius muscle, which is a muscle that X it innervates. -

Developmental Requirement of Gp130 Signaling in Neuronal Survival and Astrocyte Differentiation
The Journal of Neuroscience, July 1, 1999, 19(13):5429–5434 Developmental Requirement of gp130 Signaling in Neuronal Survival and Astrocyte Differentiation Kinichi Nakashima,1,2 Stefan Wiese,3 Makoto Yanagisawa,1 Hirokazu Arakawa,1 Naoki Kimura,1 Tatsuhiro Hisatsune,4 Kanji Yoshida,5 Tadamitsu Kishimoto,6 Michael Sendtner,3 and Tetsuya Taga1 1Department of Molecular Cell Biology and 2Cell Fate Modulation Research Unit, Medical Research Institute, Tokyo Medical and Dental University, Tokyo, 101–0062, Japan, 3Clinical Research Unit for Neuroregeneration, Department of Neurology, University of Wu¨ rzburg, Wu¨ rzburg, 97080, Germany, 4Division of Integrated Biosciences, Graduate School for Frontier Science, The University of Tokyo, Tokyo, 113–8657, Japan, and 5Department of Molecular Immunology, Research Institute for Microbial Diseases, 6Osaka University, Osaka, 565–0871, Japan gp130 is a signal-transducing receptor component used in loss was detectable on day 14.5, suggesting a physiological common by the interleukin-6 (IL-6) family of hematopoietic and role of gp130 in supporting newly generated neurons during the neurotrophic cytokines, including IL-6, IL-11, leukemia- late phase of development when naturally occurring cell death inhibitory factor, ciliary neurotrophic factor, oncostatin-M, and takes place. Moreover, expression of an astrocyte marker, cardiotrophin-1. We have examined in this study a role of gp130 GFAP, was severely reduced in the brain of gp130 2/2 mice. in the nervous system by analyzing developmental cell death of Our data demonstrate that gp130 expression is essential for several neuronal populations and the differentiation of astro- survival of subgroups of differentiated motor and sensory neu- cytes in gp130-deficient mice. -

Bioengineered Model of the Human Motor Unit with Physiologically Functional Neuromuscular Junctions Rowan P
www.nature.com/scientificreports OPEN Bioengineered model of the human motor unit with physiologically functional neuromuscular junctions Rowan P. Rimington*, Jacob W. Fleming, Andrew J. Capel, Patrick C. Wheeler & Mark P. Lewis Investigations of the human neuromuscular junction (NMJ) have predominately utilised experimental animals, model organisms, or monolayer cell cultures that fail to represent the physiological complexity of the synapse. Consequently, there remains a paucity of data regarding the development of the human NMJ and a lack of systems that enable investigation of the motor unit. This work addresses this need, providing the methodologies to bioengineer 3D models of the human motor unit. Spheroid culture of iPSC derived motor neuron progenitors augmented the transcription of OLIG2, ISLET1 and SMI32 motor neuron mRNAs ~ 400, ~ 150 and ~ 200-fold respectively compared to monolayer equivalents. Axon projections of adhered spheroids exceeded 1000 μm in monolayer, with transcription of SMI32 and VACHT mRNAs further enhanced by addition to 3D extracellular matrices in a type I collagen concentration dependent manner. Bioengineered skeletal muscles produced functional tetanic and twitch profles, demonstrated increased acetylcholine receptor (AChR) clustering and transcription of MUSK and LRP4 mRNAs, indicating enhanced organisation of the post- synaptic membrane. The number of motor neuron spheroids, or motor pool, required to functionally innervate 3D muscle tissues was then determined, generating functional human NMJs that evidence pre- and post-synaptic membrane and motor nerve axon co-localisation. Spontaneous fring was signifcantly elevated in 3D motor units, confrmed to be driven by the motor nerve via antagonistic inhibition of the AChR. Functional analysis outlined decreased time to peak twitch and half relaxation times, indicating enhanced physiology of excitation contraction coupling in innervated motor units.