Gamma Motor Neurons Survive and Exacerbate Alpha Motor Neuron Degeneration In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

How Degeneration of Cells Surrounding Motoneurons Contributes to Amyotrophic Lateral Sclerosis
cells Review How Degeneration of Cells Surrounding Motoneurons Contributes to Amyotrophic Lateral Sclerosis Roxane Crabé 1, Franck Aimond 1, Philippe Gosset 1 , Frédérique Scamps 1 and Cédric Raoul 1,2,* 1 The Neuroscience Institute of Montpellier, INSERM, UMR1051, University of Montpellier, 34091 Montpellier, France; [email protected] (R.C.); [email protected] (F.A.); [email protected] (P.G.); [email protected] (F.S.) 2 Laboratory of Neurobiology, Kazan Federal University, 420008 Kazan, Russia * Correspondence: [email protected] Received: 2 October 2020; Accepted: 24 November 2020; Published: 27 November 2020 Abstract: Amyotrophic lateral sclerosis (ALS) is a fatal neurological disorder characterized by the progressive degeneration of upper and lower motoneurons. Despite motoneuron death being recognized as the cardinal event of the disease, the loss of glial cells and interneurons in the brain and spinal cord accompanies and even precedes motoneuron elimination. In this review, we provide striking evidence that the degeneration of astrocytes and oligodendrocytes, in addition to inhibitory and modulatory interneurons, disrupt the functionally coherent environment of motoneurons. We discuss the extent to which the degeneration of glial cells and interneurons also contributes to the decline of the motor system. This pathogenic cellular network therefore represents a novel strategic field of therapeutic investigation. Keywords: amyotrophic lateral sclerosis; spinal cord; cortex; motoneuron; astrocytes; interneuron; oligodendrocytes; degeneration 1. Introduction Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by the selective and progressive loss of upper and lower motoneurons. ALS begins with focal muscle weakness and wasting that relentlessly spreads to other body parts, leading to death mostly from respiratory failure within three years of onset. -

Neural Control of Movement: Motor Neuron Subtypes, Proprioception and Recurrent Inhibition
List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I Enjin A, Rabe N, Nakanishi ST, Vallstedt A, Gezelius H, Mem- ic F, Lind M, Hjalt T, Tourtellotte WG, Bruder C, Eichele G, Whelan PJ, Kullander K (2010) Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as mark- ers for fast motor neurons and partition cells. J Comp Neurol 518:2284-2304. II Wootz H, Enjin A, Wallen-Mackenzie Å, Lindholm D, Kul- lander K (2010) Reduced VGLUT2 expression increases motor neuron viability in Sod1G93A mice. Neurobiol Dis 37:58-66 III Enjin A, Leao KE, Mikulovic S, Le Merre P, Tourtellotte WG, Kullander K. 5-ht1d marks gamma motor neurons and regulates development of sensorimotor connections Manuscript IV Enjin A, Leao KE, Eriksson A, Larhammar M, Gezelius H, Lamotte d’Incamps B, Nagaraja C, Kullander K. Development of spinal motor circuits in the absence of VIAAT-mediated Renshaw cell signaling Manuscript Reprints were made with permission from the respective publishers. Cover illustration Carousel by Sasha Svensson Contents Introduction.....................................................................................................9 Background...................................................................................................11 Neural control of movement.....................................................................11 The motor neuron.....................................................................................12 Organization -

Inside Your Brain You and Your Brain
Inside your brain You and your brain Many simple and complex psychological functions are mediated by multiple brain regions and, at the same time, a single brain area may control many psychological functions. CC BY Illustration by Bret Syfert 1. Cortex: The thin, folded structure on the outside surface of the brain. 2. Cerebral hemispheres: The two halves of the brain, each of which controls and receives information from the opposite side of the body. 3. Pituitary gland: The ‘master gland’ of the body, which releases hormones that control growth, blood pressure, the stress response and the function of the sex organs. 4. Substantia nigra: The ‘black substance’ contains cells that produce the neurotransmitter dopamine and the pigment melatonin, giving it a black appearance. 5. Hypothalamus: The interface between the brain and pituitary gland. It controls the production and release of hormones. 6. Spinal cord: A large bundle of millions of nerve fibres and neuronal cells, which carries information back and forth between the brain and the body. 7. Medulla oblongata: Controls vital involuntary functions such as breathing and heart rate. 8. Cerebellum: The ‘little brain’ that controls balance and coordinates movements. It’s normally required for learning motor skills, such as riding a bike, and is involved in thought processes. 9. Cranial nerve nuclei: Clusters of neurons in the brain stem. Their axons form the cranial nerves. Your brain underpins who you are. It stores your knowledge and memories, gives you the capacity for thought and emotion, and enables you to control your body. The brain is just one part of the nervous system. -

Speed and Segmentation Control Mechanisms Characterized in Rhythmically- Active Circuits Created from Spinal Neurons Produced Fr
RESEARCH ARTICLE Speed and segmentation control mechanisms characterized in rhythmically- active circuits created from spinal neurons produced from genetically-tagged embryonic stem cells Matthew J Sternfeld1,2, Christopher A Hinckley1, Niall J Moore1, Matthew T Pankratz1, Kathryn L Hilde1,3, Shawn P Driscoll1, Marito Hayashi1,2, Neal D Amin1,3,4, Dario Bonanomi1†, Wesley D Gifford1,4,5, Kamal Sharma6, Martyn Goulding7, Samuel L Pfaff1* 1Gene Expression Laboratory, Howard Hughes Medical Institute, Salk Institute for Biological Studies, La Jolla, United States; 2Biological Sciences Graduate Program, University of California, San Diego, La Jolla, United States; 3Biomedical Sciences Graduate Program, University of California, San Diego, La Jolla, United States; 4Medical Scientist Training Program, University of California, San Diego, La Jolla, United States; 5Neurosciences Graduate Program, University of California, San Diego, La Jolla, United States; 6Department of Anatomy and Cell Biology, University of Illinois at Chicago, Chicago, United States; 7Molecular Neurobiology Laboratory, Salk Institute for Biological Studies, La Jolla, United States *For correspondence: pfaff@salk. edu Abstract Flexible neural networks, such as the interconnected spinal neurons that control Present address: †Division of distinct motor actions, can switch their activity to produce different behaviors. Both excitatory (E) Neuroscience, San Raffaele and inhibitory (I) spinal neurons are necessary for motor behavior, but the influence of recruiting Scientific Institute, Milan, Italy different ratios of E-to-I cells remains unclear. We constructed synthetic microphysical neural networks, called circuitoids, using precise combinations of spinal neuron subtypes derived from Competing interests: The mouse stem cells. Circuitoids of purified excitatory interneurons were sufficient to generate authors declare that no oscillatory bursts with properties similar to in vivo central pattern generators. -

Deconstructing Spinal Interneurons, One Cell Type at a Time Mariano Ignacio Gabitto
Deconstructing spinal interneurons, one cell type at a time Mariano Ignacio Gabitto Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 © 2016 Mariano Ignacio Gabitto All rights reserved ABSTRACT Deconstructing spinal interneurons, one cell type at a time Mariano Ignacio Gabitto Abstract Documenting the extent of cellular diversity is a critical step in defining the functional organization of the nervous system. In this context, we sought to develop statistical methods capable of revealing underlying cellular diversity given incomplete data sampling - a common problem in biological systems, where complete descriptions of cellular characteristics are rarely available. We devised a sparse Bayesian framework that infers cell type diversity from partial or incomplete transcription factor expression data. This framework appropriately handles estimation uncertainty, can incorporate multiple cellular characteristics, and can be used to optimize experimental design. We applied this framework to characterize a cardinal inhibitory population in the spinal cord. Animals generate movement by engaging spinal circuits that direct precise sequences of muscle contraction, but the identity and organizational logic of local interneurons that lie at the core of these circuits remain unresolved. By using our Sparse Bayesian approach, we showed that V1 interneurons, a major inhibitory population that controls motor output, fractionate into diverse subsets on the basis of the expression of nineteen transcription factors. Transcriptionally defined subsets exhibit highly structured spatial distributions with mediolateral and dorsoventral positional biases. These distinctions in settling position are largely predictive of patterns of input from sensory and motor neurons, arguing that settling position is a determinant of inhibitory microcircuit organization. -

ALS and Other Motor Neuron Diseases Can Represent Diagnostic Challenges
Review Article Address correspondence to Dr Ezgi Tiryaki, Hennepin ALS and Other Motor County Medical Center, Department of Neurology, 701 Park Avenue P5-200, Neuron Diseases Minneapolis, MN 55415, [email protected]. Ezgi Tiryaki, MD; Holli A. Horak, MD, FAAN Relationship Disclosure: Dr Tiryaki’s institution receives support from The ALS Association. Dr Horak’s ABSTRACT institution receives a grant from the Centers for Disease Purpose of Review: This review describes the most common motor neuron disease, Control and Prevention. ALS. It discusses the diagnosis and evaluation of ALS and the current understanding of its Unlabeled Use of pathophysiology, including new genetic underpinnings of the disease. This article also Products/Investigational covers other motor neuron diseases, reviews how to distinguish them from ALS, and Use Disclosure: Drs Tiryaki and Horak discuss discusses their pathophysiology. the unlabeled use of various Recent Findings: In this article, the spectrum of cognitive involvement in ALS, new concepts drugs for the symptomatic about protein synthesis pathology in the etiology of ALS, and new genetic associations will be management of ALS. * 2014, American Academy covered. This concept has changed over the past 3 to 4 years with the discovery of new of Neurology. genes and genetic processes that may trigger the disease. As of 2014, two-thirds of familial ALS and 10% of sporadic ALS can be explained by genetics. TAR DNA binding protein 43 kDa (TDP-43), for instance, has been shown to cause frontotemporal dementia as well as some cases of familial ALS, and is associated with frontotemporal dysfunction in ALS. Summary: The anterior horn cells control all voluntary movement: motor activity, res- piratory, speech, and swallowing functions are dependent upon signals from the anterior horn cells. -

Lancl1 Promotes Motor Neuron Survival and Extends the Lifespan of Amyotrophic Lateral Sclerosis Mice
Cell Death & Differentiation (2020) 27:1369–1382 https://doi.org/10.1038/s41418-019-0422-6 ARTICLE LanCL1 promotes motor neuron survival and extends the lifespan of amyotrophic lateral sclerosis mice 1 1 1 1 1 1 1 Honglin Tan ● Mina Chen ● Dejiang Pang ● Xiaoqiang Xia ● Chongyangzi Du ● Wanchun Yang ● Yiyuan Cui ● 1,2 1 1,3 4 4 3 1,5 Chao Huang ● Wanxiang Jiang ● Dandan Bi ● Chunyu Li ● Huifang Shang ● Paul F. Worley ● Bo Xiao Received: 19 November 2018 / Revised: 3 September 2019 / Accepted: 6 September 2019 / Published online: 30 September 2019 © The Author(s) 2019. This article is published with open access Abstract Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive loss of motor neurons. Improving neuronal survival in ALS remains a significant challenge. Previously, we identified Lanthionine synthetase C-like protein 1 (LanCL1) as a neuronal antioxidant defense gene, the genetic deletion of which causes apoptotic neurodegeneration in the brain. Here, we report in vivo data using the transgenic SOD1G93A mouse model of ALS indicating that CNS-specific expression of LanCL1 transgene extends lifespan, delays disease onset, decelerates symptomatic progression, and improves motor performance of SOD1G93A mice. Conversely, CNS-specific deletion of fl 1234567890();,: 1234567890();,: LanCL1 leads to neurodegenerative phenotypes, including motor neuron loss, neuroin ammation, and oxidative damage. Analysis reveals that LanCL1 is a positive regulator of AKT activity, and LanCL1 overexpression restores the impaired AKT activity in ALS model mice. These findings indicate that LanCL1 regulates neuronal survival through an alternative mechanism, and suggest a new therapeutic target in ALS. -

GDE2 Regulates Subtype-Specific Motor Neuron Generation Through
Neuron Article GDE2 Regulates Subtype-Specific Motor Neuron Generation through Inhibition of Notch Signaling Priyanka Sabharwal,1 Changhee Lee,1 Sungjin Park,1 Meenakshi Rao,1,2 and Shanthini Sockanathan1,* 1The Solomon Snyder Department of Neuroscience, The Johns Hopkins University School of Medicine, PCTB1004, 725 N. Wolfe Street, Baltimore, MD 21205, USA 2Present address: Department of Pediatrics, Children’s Hospital Boston, 300 Longwood Avenue, Boston, MA 02115, USA *Correspondence: [email protected] DOI 10.1016/j.neuron.2011.07.028 SUMMARY which is innervated by specific groups of motor neurons. Indi- vidual motor neuron groups are highly organized in terms of their The specification of spinal interneuron and motor cell body distribution, projection patterns, and function and neuron identities initiates within progenitor cells, consist of force-generating alpha motor neurons that innervate while motor neuron subtype diversification is regu- extrafusal muscle fibers and stretch-sensitive gamma motor lated by hierarchical transcriptional programs imple- neurons that innervate intrafusal muscle fibers of the muscle mented postmitotically. Here we find that mice lack- spindles (Dasen and Jessell, 2009; reviewed in Kanning et al., ing GDE2, a six-transmembrane protein that triggers 2010). The integration of input from both alpha and gamma motor neurons is essential for coordinated motor movement to motor neuron generation, exhibit selective losses occur (Kanning et al., 2010). of distinct motor neuron subtypes, specifically in How is diversity engendered in developing motor neurons? defined subsets of limb-innervating motor pools All motor neurons initially derive from ventral progenitor cells that correlate with the loss of force-generating alpha that are specified to become Olig2+ motor neuron progenitors motor neurons. -

Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems
cells Review Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems Naoki Abe 1, Tasuku Nishihara 1,*, Toshihiro Yorozuya 1 and Junya Tanaka 2 1 Department of Anesthesia and Perioperative Medicine, Ehime University Graduate School of Medicine, Toon, Ehime 791-0295, Japan; [email protected] (N.A.); [email protected] (T.Y.) 2 Department of Molecular and cellular Physiology, Ehime University Graduate School of Medicine, Toon, Ehime 791-0295, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-89-960-5383; Fax: +81-89-960-5386 Received: 4 August 2020; Accepted: 17 September 2020; Published: 21 September 2020 Abstract: Microglia, the immunocompetent cells in the central nervous system (CNS), have long been studied as pathologically deteriorating players in various CNS diseases. However, microglia exert ameliorating neuroprotective effects, which prompted us to reconsider their roles in CNS and peripheral nervous system (PNS) pathophysiology. Moreover, recent findings showed that microglia play critical roles even in the healthy CNS. The microglial functions that normally contribute to the maintenance of homeostasis in the CNS are modified by other cells, such as astrocytes and infiltrated myeloid cells; thus, the microglial actions on neurons are extremely complex. For a deeper understanding of the pathophysiology of various diseases, including those of the PNS, it is important to understand microglial functioning. In this review, we discuss both the favorable and unfavorable roles of microglia in neuronal survival in various CNS and PNS disorders. We also discuss the roles of blood-borne macrophages in the pathogenesis of CNS and PNS injuries because they cooperatively modify the pathological processes of resident microglia. -

Reduction of Ephrin-A5 Aggravates Disease Progression in Amyotrophic
Rué et al. Acta Neuropathologica Communications (2019) 7:114 https://doi.org/10.1186/s40478-019-0759-6 RESEARCH Open Access Reduction of ephrin-A5 aggravates disease progression in amyotrophic lateral sclerosis Laura Rué1,2 , Patrick Oeckl3, Mieke Timmers1,2, Annette Lenaerts1,2, Jasmijn van der Vos1,2, Silke Smolders1,2, Lindsay Poppe1,2, Antina de Boer1,2, Ludo Van Den Bosch1,2, Philip Van Damme1,2,4, Jochen H. Weishaupt3, Albert C. Ludolph3, Markus Otto3, Wim Robberecht1,4 and Robin Lemmens1,2,4* Abstract Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that affects motor neurons in the brainstem, spinal cord and motor cortex. ALS is characterized by genetic and clinical heterogeneity, suggesting the existence of genetic factors that modify the phenotypic expression of the disease. We previously identified the axonal guidance EphA4 receptor, member of the Eph-ephrin system, as an ALS disease-modifying factor. EphA4 genetic inhibition rescued the motor neuron phenotype in zebrafish and a rodent model of ALS. Preventing ligands from binding to the EphA4 receptor also successfully improved disease, suggesting a role for EphA4 ligands in ALS. One particular ligand, ephrin-A5, is upregulated in reactive astrocytes after acute neuronal injury and inhibits axonal regeneration. Moreover, it plays a role during development in the correct pathfinding of motor axons towards their target limb muscles. We hypothesized that a constitutive reduction of ephrin-A5 signalling would benefit disease progression in a rodent model for ALS. We discovered that in the spinal cord of control and symptomatic ALS mice ephrin-A5 was predominantly expressed in neurons. -
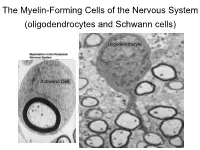
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Cortex Brainstem Spinal Cord Thalamus Cerebellum Basal Ganglia
Harvard-MIT Division of Health Sciences and Technology HST.131: Introduction to Neuroscience Course Director: Dr. David Corey Motor Systems I 1 Emad Eskandar, MD Motor Systems I - Muscles & Spinal Cord Introduction Normal motor function requires the coordination of multiple inter-elated areas of the CNS. Understanding the contributions of these areas to generating movements and the disturbances that arise from their pathology are important challenges for the clinician and the scientist. Despite the importance of diseases that cause disorders of movement, the precise function of many of these areas is not completely clear. The main constituents of the motor system are the cortex, basal ganglia, cerebellum, brainstem, and spinal cord. Cortex Basal Ganglia Cerebellum Thalamus Brainstem Spinal Cord In very broad terms, cortical motor areas initiate voluntary movements. The cortex projects to the spinal cord directly, through the corticospinal tract - also known as the pyramidal tract, or indirectly through relay areas in the brain stem. The cortical output is modified by two parallel but separate re entrant side loops. One loop involves the basal ganglia while the other loop involves the cerebellum. The final outputs for the entire system are the alpha motor neurons of the spinal cord, also called the Lower Motor Neurons. Cortex: Planning and initiation of voluntary movements and integration of inputs from other brain areas. Basal Ganglia: Enforcement of desired movements and suppression of undesired movements. Cerebellum: Timing and precision of fine movements, adjusting ongoing movements, motor learning of skilled tasks Brain Stem: Control of balance and posture, coordination of head, neck and eye movements, motor outflow of cranial nerves Spinal Cord: Spontaneous reflexes, rhythmic movements, motor outflow to body.