ALS and Other Motor Neuron Diseases Can Represent Diagnostic Challenges
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinically Undetected Motor Neuron Disease in Pathologically Proven Frontotemporal Lobar Degeneration with Motor Neuron Disease
ORIGINAL CONTRIBUTION Clinically Undetected Motor Neuron Disease in Pathologically Proven Frontotemporal Lobar Degeneration With Motor Neuron Disease Keith A. Josephs, MST, MD; Joseph E. Parisi, MD; David S. Knopman, MD; Bradley F. Boeve, MD; Ronald C. Petersen, MD, PhD; Dennis W. Dickson, MD Background: Frontotemporal lobar degeneration with evidence of motor neuron disease. Semiquantitative motor neuron disease (FTLD-MND) is a pathological analysis of motor and extramotor pathological findings entity characterized by motor neuron degeneration and revealed a spectrum of pathological changes underlying frontotemporal lobar degeneration. The ability to detect FTLD-MND. Hippocampal sclerosis, predominantly of the clinical signs of dementia and motor neuron disease the subiculum, was a significantly more frequent occur- in pathologically confirmed FTLD-MND has not been rence in the cases without clinical evidence of motor assessed. neuron disease (PϽ.01). In addition, neuronal loss, gliosis, and corticospinal tract degeneration were less Objectives: To determine if all cases of pathologically severe in the other 3 cases without clinical evidence of confirmed FTLD-MND have clinical evidence of fronto- motor neuron disease. temporal dementia and motor neuron disease, and to de- termine the possible reasons for misdiagnosis. Conclusions: Clinical diagnostic sensitivity for the el- ements of FTLD-MND is modest and may be affected by Method: Review of historical records and semiquantita- the fact that FTLD-MND represents a spectrum of patho- tive analysis of the motor and extramotor pathological find- logical findings, rather than a single homogeneous en- ings of all cases of pathologically confirmed FTLD-MND. tity. Detection of signs of clinical motor neuron disease is also difficult when motor neuron degeneration is mild Results: From a total of 17 cases of pathologically con- and in patients with hippocampal sclerosis. -

Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia
brain sciences Review Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia Timothy Fullam and Jeffrey Statland * Department of Neurology, University of Kansas Medical Center, Kansas, KS 66160, USA; [email protected] * Correspondence: [email protected] Abstract: Following the exclusion of potentially reversible causes, the differential for those patients presenting with a predominant upper motor neuron syndrome includes primary lateral sclerosis (PLS), hereditary spastic paraplegia (HSP), or upper motor neuron dominant ALS (UMNdALS). Differentiation of these disorders in the early phases of disease remains challenging. While no single clinical or diagnostic tests is specific, there are several developing biomarkers and neuroimaging technologies which may help distinguish PLS from HSP and UMNdALS. Recent consensus diagnostic criteria and use of evolving technologies will allow more precise delineation of PLS from other upper motor neuron disorders and aid in the targeting of potentially disease-modifying therapeutics. Keywords: primary lateral sclerosis; amyotrophic lateral sclerosis; hereditary spastic paraplegia Citation: Fullam, T.; Statland, J. Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper 1. Introduction Motor Neuron Dominant Jean-Martin Charcot (1825–1893) and Wilhelm Erb (1840–1921) are credited with first Amyotrophic Lateral Sclerosis, and describing a distinct clinical syndrome of upper motor neuron (UMN) tract degeneration in Hereditary Spastic Paraplegia. Brain isolation with symptoms including spasticity, hyperreflexia, and mild weakness [1,2]. Many Sci. 2021, 11, 611. https:// of the earliest described cases included cases of hereditary spastic paraplegia, amyotrophic doi.org/10.3390/brainsci11050611 lateral sclerosis, and underrecognized structural, infectious, or inflammatory etiologies for upper motor neuron dysfunction which have since become routinely diagnosed with the Academic Editors: P. -

Neural Control of Movement: Motor Neuron Subtypes, Proprioception and Recurrent Inhibition
List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I Enjin A, Rabe N, Nakanishi ST, Vallstedt A, Gezelius H, Mem- ic F, Lind M, Hjalt T, Tourtellotte WG, Bruder C, Eichele G, Whelan PJ, Kullander K (2010) Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as mark- ers for fast motor neurons and partition cells. J Comp Neurol 518:2284-2304. II Wootz H, Enjin A, Wallen-Mackenzie Å, Lindholm D, Kul- lander K (2010) Reduced VGLUT2 expression increases motor neuron viability in Sod1G93A mice. Neurobiol Dis 37:58-66 III Enjin A, Leao KE, Mikulovic S, Le Merre P, Tourtellotte WG, Kullander K. 5-ht1d marks gamma motor neurons and regulates development of sensorimotor connections Manuscript IV Enjin A, Leao KE, Eriksson A, Larhammar M, Gezelius H, Lamotte d’Incamps B, Nagaraja C, Kullander K. Development of spinal motor circuits in the absence of VIAAT-mediated Renshaw cell signaling Manuscript Reprints were made with permission from the respective publishers. Cover illustration Carousel by Sasha Svensson Contents Introduction.....................................................................................................9 Background...................................................................................................11 Neural control of movement.....................................................................11 The motor neuron.....................................................................................12 Organization -

Approach to a Patient with Hemiplegia and Monoplegia
CHAPTER Approach to a Patient with Hemiplegia and Monoplegia 27 Sudhir Kumar, Subhash Kaul INTRODUCTION 4. Injury to multiple cervical nerve roots. Monoplegia and hemiplegia are common neurological 5. Functional or psychogenic. symptoms in patients presenting to the emergency department as well as outpatient department. Insidious onset, gradually progressive monoplegia affecting lower limb can be caused by the following Monoplegia refers to weakness of one limb (either arm or conditions: leg) and hemiplegia refers to weakness of one arm and leg on the same side of body (either left or right side). 1. Tumor of the contralateral frontal lobe. There are a variety of underlying causes for monoplegia 2. Tumor of spinal cord at thoracic or lumbar level. and hemiplegia. The causes differ in different age groups. 3. Chronic infection of brain (frontal lobe) or spinal The causes also differ depending on the onset, progression cord (thoracic or lumbar level), such as tuberculous. and duration of weakness. Therefore, one needs to adopt a systematic approach during history taking and 4. Lumbosacral-plexopathy, due to diabetes mellitus. examination in order to arrive at the correct diagnosis. Insidious onset, gradually progressive monoplegia, Appropriate investigations after these would confirm the affecting upper limb, can be caused by one of the following diagnosis. conditions: The aim of this chapter is to systematically look at the 1. Tumor of the contralateral parietal lobe. differential diagnosis of monoplegia and hemiplegia and outline the approach needed to pinpoint the exact 2. Compressive lesion (tumor, large disc, etc) in underlying cause. cervical cord region. 3. Chronic infection of the brain (parietal lobe) or APPROACH TO THE DIAGNOSIS OF MONOPLEGIA spinal cord (cervical region), such as tuberculous. -

Neuromuscular Disorders Neurology in Practice: Series Editors: Robert A
Neuromuscular Disorders neurology in practice: series editors: robert a. gross, department of neurology, university of rochester medical center, rochester, ny, usa jonathan w. mink, department of neurology, university of rochester medical center,rochester, ny, usa Neuromuscular Disorders edited by Rabi N. Tawil, MD Professor of Neurology University of Rochester Medical Center Rochester, NY, USA Shannon Venance, MD, PhD, FRCPCP Associate Professor of Neurology The University of Western Ontario London, Ontario, Canada A John Wiley & Sons, Ltd., Publication This edition fi rst published 2011, ® 2011 by Blackwell Publishing Ltd Blackwell Publishing was acquired by John Wiley & Sons in February 2007. Blackwell’s publishing program has been merged with Wiley’s global Scientifi c, Technical and Medical business to form Wiley-Blackwell. Registered offi ce: John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK Editorial offi ces: 9600 Garsington Road, Oxford, OX4 2DQ, UK The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK 111 River Street, Hoboken, NJ 07030-5774, USA For details of our global editorial offi ces, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com/wiley-blackwell The right of the author to be identifi ed as the author of this work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. -

Motor Neuron Disease and the Elderly
Neurology 61 Motor neuron disease and the elderly Motor neuron disease is a devastating condition characterised by degeneration of motor nerves. Many of the presenting symptoms, such as fatigue, muscle weakness and difficulty in swallowing have a broad differential diagnoses in the elderly population. Dr Sheba Azam and Professor PN Leigh explain how ensuring quality of life for patients requires preventing unnecessary delay in diagnosis and early referral to an appropriate multidisciplinary team. myotrophic lateral sclerosis (ALS) also cognitive changes. Thus, misdiagnosis as well as known as motor neuron disease (MND) under-investigation has been suggested as possible (the terms are used interchangeably), was causes of an apparent decrease in the incidence of A 4 fi rst described in 1869 by the French neurologist MND in later life . Jean-Martin Charcot1. It is a progressive, fatal neurological disease characterised by degeneration of motor nerve cells in the motor cortex, Prognostic factors corticospinal tract and the spinal cord anterior horn Although the average survival in MND is around cells. The degeneration of motor nerve cells results 36 months, some patients live for 10 years or more. in progressive muscle wasting leading to signifi cant Certain phenotypic variants appear to determine disability and ultimately death. Death usually survival rates. Using information held in a tertiary results from respiratory failure due to weakness of referral MND database, a group of researchers the respiratory muscles. analysed data on onset of disease, site of onset and duration of survival5. The authors concluded that typical MND with bulbar onset, onset later in life Incidence and prevalence or in the defi nite category of El Escorial (where The worldwide incidence of MND is approximately the World Federation of Neurologist meet to a professor of clinical two per 100,000 and the prevalence is four to seven decide diagnostic criteria) at presentation, per 100,000. -

A System for Studying Mechanisms of Neuromuscular Junction Development and Maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 2464-2477 doi:10.1242/dev.130278 TECHNIQUES AND RESOURCES RESEARCH ARTICLE A system for studying mechanisms of neuromuscular junction development and maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R. Gomes1,3,*,‡ ABSTRACT different animal models and cell lines (Chen et al., 2014; Corti et al., The neuromuscular junction (NMJ), a cellular synapse between a 2012; Lenzi et al., 2015) with the hope of recapitulating some motor neuron and a skeletal muscle fiber, enables the translation of features of neuromuscular diseases and understanding the triggers chemical cues into physical activity. The development of this special of one of their common hallmarks: the disruption of the structure has been subject to numerous investigations, but its neuromuscular junction (NMJ). The NMJ is one of the most complexity renders in vivo studies particularly difficult to perform. studied synapses. It is formed of three key elements: the lower motor In vitro modeling of the neuromuscular junction represents a powerful neuron (the pre-synaptic compartment), the skeletal muscle (the tool to delineate fully the fine tuning of events that lead to subcellular post-synaptic compartment) and the Schwann cell (Sanes and specialization at the pre-synaptic and post-synaptic sites. Here, we Lichtman, 1999). The NMJ is formed in a step-wise manner describe a novel heterologous co-culture in vitro method using rat following a series of cues involving these three cellular components spinal cord explants with dorsal root ganglia and murine primary and its role is basically to ensure the skeletal muscle functionality. -

Exacerbation of Motor Neuron Disease by Chronic Stimulation of Innate Immunity in a Mouse Model of Amyotrophic Lateral Sclerosis
1340 • The Journal of Neuroscience, February 11, 2004 • 24(6):1340–1349 Neurobiology of Disease Exacerbation of Motor Neuron Disease by Chronic Stimulation of Innate Immunity in a Mouse Model of Amyotrophic Lateral Sclerosis Minh Dang Nguyen,1 Thierry D’Aigle,2 Genevie`ve Gowing,1,2 Jean-Pierre Julien,1,2 and Serge Rivest2 1McGill University Health Center, Centre for Research in Neurosciences, McGill University, The Montreal General Hospital Research Institute, Montre´al, Que´bec H3G 1A4, Canada, and 2Laboratory of Molecular Endocrinology, Laval University Medical Center Research Center and Department of Anatomy and Physiology, Laval University, Sainte-Foy, Que´bec G1V 4G2, Canada Innate immunity is a specific and organized immunological program engaged by peripheral organs and the CNS to maintain homeostasis after stress and injury. In neurodegenerative disorders, its putative deregulation, featured by inflammation and activation of glial cells resulting from inherited mutations or viral/bacterial infections, likely contributes to neuronal death. However, it remains unclear to what extent environmental factors and innate immunity cooperate to modulate the interactions between the neuronal and non-neuronal elements in the perturbed CNS. In the present study, we addressed the effects of acute and chronic administration of lipopolysaccharide (LPS), a Gram-negative bacterial wall component, in a genetic model of neurodegeneration. Transgenic mice expressing a mutant form of the superoxide dismutase 1 (SOD1 G37R) linked to familial amyotrophic lateral sclerosis were challenged intraperitoneally with a single nontoxic or repeated injections of LPS (1 mg/kg). At different ages, SOD1 G37R mice responded normally to acute endotoxemia. Remark- ably, only a chronic challenge with LPS in presymptomatic 6-month-old SOD1 G37R mice exacerbated disease progression by 3 weeks and motor axon degeneration. -

Cranial Nerves 1, 5, 7-12
Cranial Nerve I Olfactory Nerve Nerve fiber modality: Special sensory afferent Cranial Nerves 1, 5, 7-12 Function: Olfaction Remarkable features: – Peripheral processes act as sensory receptors (the other special sensory nerves have separate Warren L Felton III, MD receptors) Professor and Associate Chair of Clinical – Primary afferent neurons undergo continuous Activities, Department of Neurology replacement throughout life Associate Professor of Ophthalmology – Primary afferent neurons synapse with secondary neurons in the olfactory bulb without synapsing Chair, Division of Neuro-Ophthalmology first in the thalamus (as do all other sensory VCU School of Medicine neurons) – Pathways to cortical areas are entirely ipsilateral 1 2 Crania Nerve I Cranial Nerve I Clinical Testing Pathology Anosmia, hyposmia: loss of or impaired Frequently overlooked in neurologic olfaction examination – 1% of population, 50% of population >60 years Aromatic stimulus placed under each – Note: patients with bilateral anosmia often report nostril with the other nostril occluded, eg impaired taste (ageusia, hypogeusia), though coffee, cloves, or soap taste is normal when tested Note that noxious stimuli such as Dysosmia: disordered olfaction ammonia are not used due to concomitant – Parosmia: distorted olfaction stimulation of CN V – Olfactory hallucination: presence of perceived odor in the absence of odor Quantitative clinical tests are available: • Aura preceding complex partial seizures of eg, University of Pennsylvania Smell temporal lobe origin -

Lancl1 Promotes Motor Neuron Survival and Extends the Lifespan of Amyotrophic Lateral Sclerosis Mice
Cell Death & Differentiation (2020) 27:1369–1382 https://doi.org/10.1038/s41418-019-0422-6 ARTICLE LanCL1 promotes motor neuron survival and extends the lifespan of amyotrophic lateral sclerosis mice 1 1 1 1 1 1 1 Honglin Tan ● Mina Chen ● Dejiang Pang ● Xiaoqiang Xia ● Chongyangzi Du ● Wanchun Yang ● Yiyuan Cui ● 1,2 1 1,3 4 4 3 1,5 Chao Huang ● Wanxiang Jiang ● Dandan Bi ● Chunyu Li ● Huifang Shang ● Paul F. Worley ● Bo Xiao Received: 19 November 2018 / Revised: 3 September 2019 / Accepted: 6 September 2019 / Published online: 30 September 2019 © The Author(s) 2019. This article is published with open access Abstract Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive loss of motor neurons. Improving neuronal survival in ALS remains a significant challenge. Previously, we identified Lanthionine synthetase C-like protein 1 (LanCL1) as a neuronal antioxidant defense gene, the genetic deletion of which causes apoptotic neurodegeneration in the brain. Here, we report in vivo data using the transgenic SOD1G93A mouse model of ALS indicating that CNS-specific expression of LanCL1 transgene extends lifespan, delays disease onset, decelerates symptomatic progression, and improves motor performance of SOD1G93A mice. Conversely, CNS-specific deletion of fl 1234567890();,: 1234567890();,: LanCL1 leads to neurodegenerative phenotypes, including motor neuron loss, neuroin ammation, and oxidative damage. Analysis reveals that LanCL1 is a positive regulator of AKT activity, and LanCL1 overexpression restores the impaired AKT activity in ALS model mice. These findings indicate that LanCL1 regulates neuronal survival through an alternative mechanism, and suggest a new therapeutic target in ALS. -

Is Hirayama a Gq1b Disease?
Neurological Sciences (2019) 40:1743–1747 https://doi.org/10.1007/s10072-019-03758-x LETTER TO THE EDITOR Is Hirayama a Gq1b disease? Sezin AlpaydınBaslo1 & Mücahid Erdoğan1 & Zeynep Ezgi Balçık1 & Oya Öztürk 1 & Dilek Ataklı1 Received: 13 November 2018 /Accepted: 7 February 2019 /Published online: 23 February 2019 # Fondazione Società Italiana di Neurologia 2019 Introduction knowledge, the association between HD and anti-ganglioside antibodies has not been reported before. Hirayama disease (HD) or juvenile muscular atrophy of We herein, present a young male patient who got the diag- distal upper extremity is a rare benign disease of motor nosis of HD, with unilateral complains but bilateral asymmet- neurons that commonly affects the cervical spinal seg- rical hand muscle weakness and atrophy accompanied by bi- ments. It is more prevalent in males and mostly seen in lateral electrophysiology and typical MRI findings. His serum teens and early 20s. Slowly progressive unilateral or was strongly positive (138%) for anti-GQ1b IgG antibody that asymmetrically bilateral weakness of hands and fore- is worth to be mentioned as a notable bystander. arms is typical. Sensory disturbances, autonomic and Informed consent was obtained from the patient included in upper motor neuron signs are extremely rare [1]. As the study. the synonym Bbenign focal amyotrophy^ implies, it reaches a plateauafterafewyears.Electromyographyrevealsasymmetri- cal chronic denervation in C7, C8, and T1 myotomes. Case Preservation of C6 myotomes is remarkable. Supportive MRI findings are anterior shift of posterior dura, enlargement of epi- A 17-year-old male was admitted with a 1 year’shistoryof dural space, and venous congestion under neck flexion. -
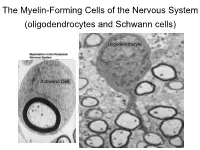
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes