4. Analytical Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

1,45%,562 UNITED SATES P All.‘ Bl If" Til?
Patented May 1, 1923. 1,45%,562 UNITED SATES P All.‘ bl if" til? . ROBERT E. WILSON, LEON ‘JV. PARSONS, AND STANLEY 1E. OHXSHOLIYI, OF WASHINGTON, DISTRICT OF COLUMBIA. PROCESS THE PRODUCTION OE‘ All‘HALLEAETELMLETAL PERTJIANGANATES. N0 Drawing". Application ?led September 27, 1918. Serial No. 255,975. To (all to]: am it may concern. .' manganate by oxidation or acidification, Be it known that we, Romain‘ E. lllinsou, metatheses into calcium pern'iangamite by LEON “7. Parsons, and STANLEY L. Unis treatment With calcium sulphate or milk at HoLM, citizens of the United States. and sta lime. tioned at ViTashington, District of Columbia, O'li' these four possible methods, (1.) is not 60 in the o?icc of the Director the Chemical a possible large scale method. on account l/Varfare Service, Research Division, have in of its use ot silver; (2) and are elec vented a Process for the ll’roduction oif Al trolytic methods Without a. great deal out kali-Earth-l\letal Permanpjanates, of which promise, and are to be considered elsewhere; ll) the ‘following is a speci?cation. (ll) the principal subject of this applica G3 in The present invention relates to the pro tion. duction oi? alkah» earth metal permangam Three distinct methods for preparing: ba~ nates and especially the permanganates of rium (or strontium) manganate have been calcium and magnesium as these have beenv here investigated. The ?rst of? these meth found to be very ellicient oxidizing agents ods involves heating together barium perox 70 for certain purposes, more e?icient even. than ide, hydroxide, or a salt, such as the nitrate the permanganates of the allmliearth metals. -

Hazardous Material Inventory Statement
City of Brooklyn Park FIRE DEPARTMENT 5200 - 85th Avenue North Brooklyn Park MN 55443 Phone: (763)493-8020 Fax: (763) 493-8391 Hazardous Materials Inventory Statement Users Guide A separate inventory statement shall be provided for each building. An amended inventory statement shall be provided within 30 days of the storage of any hazardous materials or plastics that changes or adds a hazard class or which is sufficient in quantity to cause an increase in the quantity which exceeds 5 percent for any hazard class. The hazardous materials inventory statement shall list by hazard class categories. Each grouping shall provide the following information for each hazardous material listed for that group including a total quantity for each group of hazard class. 1. Hazard class. (See attached Hazardous Materials Categories Listing) 2. Common or trade name. 3. Chemical Abstract Service Number (CAS number) found in 29 Code of Federal Regulations (C.F.R.). 4. Whether the material is pure or a mixture, and whether the material is a solid, liquid or gas 5. Maximum aggregate quantity stored at any one time. 6. Maximum aggregate quantity In-Use (Open to atmosphere) at any one time. 7. Maximum aggregate quantity In-Use (Closed to atmosphere) at any one time. 8. Storage conditions related to the storage type, high-pile, encapsulated, non-encapsulated. Attached is a listing of categories that all materials need to be organized to. Definitions of these categories are also attached for your use. At the end of this packet are blank forms for completing this project. For questions regarding Hazardous Materials Inventory Statement contact the Fire Department at 763-493-8020. -

Prohibited and Restricted Chemical List
School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List PROHIBITED AND RESTRICTED CHEMICAL LIST Introduction After incidents of laboratory chemical contamination at several schools, DCPS, The American Association for the Advancement of Science (AAAS) and DC Fire and Emergency Management Services developed an aggressive program for chemical control to eliminate student and staff exposure to potential hazardous chemicals. Based upon this program, all principals are required to conduct a complete yearly inventory of all chemicals located at each school building to identify for the removal and disposal of any prohibited/banned chemicals. Prohibited chemicals are those that pose an inherent, immediate, and potentially life- threatening risk, injury, or impairment due to toxicity or other chemical properties to students, staff, or other occupants of the school. These chemicals are prohibited from use and/or storage at the school, and the school is prohibited from purchasing or accepting donations of such chemicals. Restricted chemicals are chemicals that are restricted by use and/or quantities. If restricted chemicals are present at the school, each storage location must be addressed in the school's written emergency plan. Also, plan maps must clearly denote the storage locations of these chemicals. Restricted chemicals—demonstration use only are a subclass in the Restricted chemicals list that are limited to instructor demonstration. Students may not participate in handling or preparation of restricted chemicals as part of a demonstration. If Restricted chemicals—demonstration use only are present at the school, each storage location must be addressed in the school's written emergency plan. Section 7: Appendices – October 2009 37 School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List Following is a table of chemicals that are Prohibited—banned, Restricted—academic curriculum use, and Restricted—demonstration use only. -

LABORATORY SAFETY and CHEMICAL HYGIENE PLAN
LABORATORY SAFETY and CHEMICAL HYGIENE PLAN INDIANA UNIVERSITY Produced by University Environmental Health and Safety December 1996 in cooperation with University Chemical Hygiene Committees Revised 2015 Page 1 Table of Contents EMERGENCY INFORMATION .................................................................................................. 4 Major Emergencies............................................................................................................ 4 Minor Emergencies............................................................................................................ 4 Accident Reports ............................................................................................................... 4 Fires and Fire Alarms ........................................................................................................ 5 Tornado Watches and Warnings (Sirens) .......................................................................... 6 LABORATORY SAFETY CONTACTS ...................................................................................... 7 1. INTRODUCTION ........................................................................................................10 1.1. Purpose ................................................................................................................ 10 1.2. Regulatory Basis ................................................................................................... 10 1.3. Applicability .......................................................................................................... -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
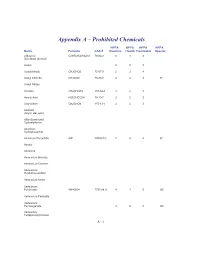
Appendix a – Prohibited Chemicals
Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special 2-Butanol C2H5CH(OH)CH3 78-92-2 0 1 3 (Sec-Butyl Alcohol) Acetal 023 Acetaldehyde CH3CHO5 75-07-0 2 3 4 Acetyl Chloride CH3COCl 75-36-5 2 3 3 W Acetyl Nitrate Acrolein CH2CHCHO 107-02-8 3 4 3 Acrylic Acid H2CCHCO2H 79-10-7 2 2 2 Acrylonitrile CH2CHCN 107-13-1 2 4 3 Alcohols (Allylic, Benzylic) Alkly-Substituted Cycloaliphatics Aluminum Hydrophosphide Aluminum Phosphide AlP 20859-73- 2 4 4 W Amatol Ammonal Ammonium Bromate Ammonium Chlorate Ammonium Hexanitrocobaltate Ammonium Nitrite Ammonium Perchlorate NH4ClO4 7790-98-9 4 1 0 OX Ammonium Periodate Ammonium Permanganate 300 OX Ammonium Tetraperoxychromate A - 1 Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special Antimony Compounds Arsenic And Arsenic Compounds Azides Azidocarbonyl Guanidine Barium Ba 2 2 1 W Barium Chlorate Ba(ClO3)2*H2O 13477-00- 1 2 0 OX Barium Oxide (Anhydrous) BaO 1304-28-5 2 3 0 Barium Peroxide BaO2 1304-29-6 0 1 0 OX Benzene C6H6 71-43-2 0 2 3 Benzene Diazonium Chloride Benzotriazole C6H5N3 95-14-7 0 2 1 Benzoyl Peroxide (C6H5CO)2O2 94-36-0 4 1 4 OX Benzyl Alcohol C6H5CH2OH 100-51-6 0 2 1 Bismuth Nitrate Bi(NO3)3*5H2O 10035-06- 3 1 0 OX Borane,Boranes, Diboranes Boron Tribromide 230 W Boron Trifluoride 140 Bromine Pentafluoride Brf5 7789-30-2 3 4 0 W,O Bromine Trifluoride 3 4 0 W,O Butadiene C4H6/CH2=(CH)2=CH 106-99-0 0 2 4 Butenetroil Trinitrate Cadmium and Cadmium Compounds Calcium Nitrate, Anhydrous Ca(NO3)2 -

Fedex Ground Hazardous Materials Shipping Guide Is Intended to Simplify Title 49 CFR
FedEx Ground Package Systems Inc. is committed to the safe transportation of hazardous materials. It is very important that each person engaged in the transportation of hazardous materials has the proper training and is thoroughly familiar with the Title 49CFR (Code of Federal Regulations) and/or USPS Publication 52. This guide is intended only to assist you in your preparation of hazardous materials shipped via FedEx Ground Package Systems Inc. It is the shipper’s responsibility to ensure each hazardous material package is in compliance with applicable Department of Transportation (D.O.T.) regulations and FedEx Ground Package Systems Inc. requirements. Failure to comply with these regulations and requirements may subject the shipper and carrier to fines and penalties. Improperly prepared hazmat packages or documentation may be subject to an additional charge(s) due to the unexpected hanlding associated with these shipments. Due to the changing nature of D.O.T. regulations and other information, it is impossible to guarantee absolute accuracy of the material contained in this guide. FedEx Ground Package Systems Inc., therefore, cannot assume any responsibility for omissions, errors, misprinting, or ambiguity contained within this guide and shall not be held liable in any degree for any loss or injury caused by such omission or error presented in this publication. Shippers should consult the most current version of the hazardous material regulations. Training is mandatory for those shipping hazardous materials, including limited quantity and other exceptions. The www.shipsafeshipsmart.com battery and hazmat training programs offer shippers an economical source of basic ground battery and/or hazardous materials shipping as well as addressing FedEx Ground specific issues. -

Standard X-Ray Diffraction Powder Patterns NATIONAL BUREAU of STANDARDS
NBS MONOGRAPH 25—SECTION 1 9 CO Q U.S. DEPARTMENT OF COMMERCE/National Bureau of Standards Standard X-ray Diffraction Powder Patterns NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress on March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau's technical work is per- formed by the National Measurement Laboratory, the National Engineering Laboratory, and the Institute for Computer Sciences and Technology. THE NATIONAL MEASUREMENT LABORATORY provides the national system of physical and chemical and materials measurement; coordinates the system with measurement systems of other nations and furnishes essentia! services leading to accurate and uniform physical and chemical measurement throughout the Nation's scientific community, industry, and commerce; conducts materials research leading to improved methods of measurement, standards, and data on the properties of materials needed by industry, commerce, educational institutions, and Government; provides advisory and research services to other Government agencies; develops, produces, and distributes Standard Reference Materials; and provides calibration -

Precipitation of Enriched Lutetium by Direct Oxalate Extraction
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Supervised Undergraduate Student Research Chancellor’s Honors Program Projects and Creative Work Spring 5-1999 Precipitation of Enriched Lutetium by Direct Oxalate Extraction Paul Dennis Campbell University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_chanhonoproj Recommended Citation Campbell, Paul Dennis, "Precipitation of Enriched Lutetium by Direct Oxalate Extraction" (1999). Chancellor’s Honors Program Projects. https://trace.tennessee.edu/utk_chanhonoproj/290 This is brought to you for free and open access by the Supervised Undergraduate Student Research and Creative Work at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Chancellor’s Honors Program Projects by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. UNIVERSITY HONORS PROGRAM SENIOR PROJECT. APPROVAL Name: "l.Il ..D.. Caifll>l>el \ .,Jr. ________________________________ _ f ~-- ----+---~------ College: B!.+l_q[ll_iE!!~~________ Department: E-_~.!..f:!.'t _______________ _ Faculty Mentor: Jd~_~!2!"'!:_t..:_~~~~i.t~.!: _________________________ _ PROJECT TITLE: _er:t~t.!J.!h·~ _ _95_§i1d~~cf.._fyAhjJ~_§t_~~!.d._Q~a~~~fK'!'i.c!{~_ ---------------------------------------------------------- I have review is completed senior honors thesis with this student and certify that it is a pr commen e with honors level undergraduate research in this field. Si gned: ____.!._-t:.''- _______+ _____________ ,F acu Ity Men tor Date: --~.l_M~-j!t.f.2-- Comments (Optional): -- Precipitation of Enriched Lutetium by Direct Oxalate Extraction A Senior Honors Paper by Paul Dennis Campbell, Jr. Experimentation by Paul D. -

Europium, Yttrium, and Indium Recovery from Electronic Wastes
metals Article Europium, Yttrium, and Indium Recovery from Electronic Wastes Ernesto de la Torre *, Estefanía Vargas, César Ron and Sebastián Gámez Department of Extractive Metallurgy, Escuela Politécnica Nacional, Ladrón de Guevara E11-253, Quito 170517, Ecuador; [email protected] (E.V.); [email protected] (C.R.); [email protected] (S.G.) * Correspondence: [email protected]; Tel.: +593-(9)9947-1051 Received: 15 September 2018; Accepted: 27 September 2018; Published: 29 September 2018 Abstract: Waste electrical and electronic equipment (WEEE) has increased in recent decades due to the continuous advancement of technology in the modern world. These residues have various metals that are found in concentrations that make their recovery profitable. A group of metals of interest are the rare earths such as europium and yttrium, as well as semiconductors such as indium. Yttrium was recovered from cathode ray tubes that were manually dismantled. The resulted powder was leached with HNO3, and then the solution was submitted to solvent extraction with di-(2-ethylhexyl) phosphoric acid (DEHPA) using n-heptane as a diluent. For re-extraction, HNO3 was used again, and yttrium was precipitated by adding four times the stoichiometric amount of oxalic acid, reaching 68% yttrium purity. Indium was recovered from the liquid crystal display (LCD) screens for which the pulverized material was leached with H2SO4. Then, the indium sulfate was subjected to solvent extraction using DEHPA as an extractant, and diesel as a diluent. The re-extraction was carried out again with H2SO4, and the obtained acid solution was evaporated until the indium precipitated, reaching a recovery of 95%. -

Hazardous Waste List (California Code of Regulations, Title 22 Section 66261.126)
Hazardous Waste List (California Code of Regulations, Title 22 Section 66261.126) Appendix X - List of Chemical Names and Common Names for Hazardous Wastes and Hazardous Materials (a) This subdivision sets forth a list of chemicals which create a presumption that a waste is a hazardous waste. If a waste consists of or contains a chemical listed in this subdivision, the waste is presumed to be a hazardous waste Environmental Regulations of CALIFORNIA unless it is determined that the waste is not a hazardous waste pursuant to the procedures set forth in section 66262.11. The hazardous characteristics which serve as a basis for listing the chemicals are indicated in the list as follows: (X) toxic (C) corrosive (I) ignitable (R) reactive * =Extremely Hazardous A chemical denoted with an asterisk is presumed to be an extremely hazardous waste unless it does not exhibit any of the criteria set forth in section 66261.110 and section 66261.113. Trademark chemical names are indicated by all capital letters. 1. Acetaldehyde (X,I) 2. Acetic acid (X,C,I) 3. Acetone, Propanone (I) 4. *Acetone cyanohydrin (X) 5. Acetonitrile (X,I) 6. *2-Acetylaminofluorene, 2-AAF (X) 7. Acetyl benzoyl peroxide (X,I,R) 8. *Acetyl chloride (X,C,R) 9. Acetyl peroxide (X,I,R) 10. Acridine (X) 11. *Acrolein, Aqualin (X,I) 12. *Acrylonitrile (X,I) 13. *Adiponitrile (X) 14. *Aldrin; 1,2,3,4,10,10-Hexachloro-1,4,4a,5,8,8a-hexahydro-1,4,5,8-endo-exodimethanonaphthlene (X) 15. *Alkyl aluminum chloride (C,I,R) 16. *Alkyl aluminum compounds (C,I,R) 17.