The Formation and Reactivitt of Some Transition Metal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effect of Mercury on Engineering Materials Used in Ammonia Plants
Effect of Mercury on Engineering Materials Used in Ammonia Plants Inappropriate combinations of moisture, alloy and mercury can accelerate corrosion and form explosive compounds. Are there ways of preventing these potential hazards? S.Mark Wilhelm Cortest Laboratories, Inc., Cypress, TX 77429 INTRODUCTION Secondly, concentration of mercury in LNG and ethylene plants has Elemental mercury (Hg) is becoming presented a range of materials an increasingly more prevalent related problems having to do with contaminant to natural gas feed aluminum. Lastly, reaction of stocks due to its production from mercury with ammonia (or NH3 various reservoirs around the world. precursors) can deposit potentially Table 1 shows the origin of gas explosive compounds in NH3 process having mercury contamination from a equipment. variety of locations(1>. This paper discusses the following In general, mercury contamination of points relating to mercury and its gas streams does not cause major influence on gas processing and use: operational problems for gas production, transportation or 1. Origin and composition of processing, at least from the mercury and mercury compounds standpoint of engineering. There in natural gas. are, however, environmental concerns from end use"'. Environmental issues Materials degradation will not be addressed in the present mechanisms. discussion. 3. Overview of mercury related Several situations have come to equipment failures. notice since 1975 that cause concern for the gas industry as they relate 4. Mercury - nitrogen compounds. to mercury contaminated gas. One such problem is that minuscule 5. Recommendations for ammonia amounts of mercury in gas can induce plant operations. liquid metal cracking of a small group of susceptible alloys. -

148 Subpart A—General Subpart B—Table of Hazardous Materials
§ 172.1 49 CFR Ch. I (10–1–15 Edition) 172.510 Special placarding provisions: Rail. APPENDIX B TO PART 172—TREFOIL SYMBOL 172.512 Freight containers and aircraft unit APPENDIX C TO PART 172—DIMENSIONAL SPEC- load devices. IFICATIONS FOR RECOMMENDED PLACARD 172.514 Bulk packagings. HOLDER 172.516 Visibility and display of placards. APPENDIX D TO PART 172—RAIL RISK ANAL- 172.519 General specifications for placards. YSIS FACTORS 172.521 DANGEROUS placard. AUTHORITY: 49 U.S.C. 5101–5128, 44701; 49 172.522 EXPLOSIVES 1.1, EXPLOSIVES 1.2 CFR 1.81, 1.96 and 1.97. and EXPLOSIVES 1.3 placards. 172.523 EXPLOSIVES 1.4 placard. SOURCE: Amdt. 172–29, 41 FR 15996, Apr. 15, 172.524 EXPLOSIVES 1.5 placard. 1976, unless otherwise noted. 172.525 EXPLOSIVES 1.6 placard. 172.526 [Reserved] 172.527 Background requirements for cer- Subpart A—General tain placards. 172.528 NON-FLAMMABLE GAS placard. § 172.1 Purpose and scope. 172.530 OXYGEN placard. This part lists and classifies those 172.532 FLAMMABLE GAS placard. materials which the Department has 172.536 [Reserved] designated as hazardous materials for 172.540 POISON GAS placard. purposes of transportation and pre- 172.542 FLAMMABLE placard. 172.544 COMBUSTIBLE placard. scribes the requirements for shipping 172.546 FLAMMABLE SOLID placard. papers, package marking, labeling, and 172.547 SPONTANEOUSLY COMBUSTIBLE transport vehicle placarding applicable placard. to the shipment and transportation of 172.548 DANGEROUS WHEN WET placard. those hazardous materials. 172.550 OXIDIZER placard. 172.552 ORGANIC PEROXIDE placard. [Amdt. 172–29, 41 FR 15997, Apr. -
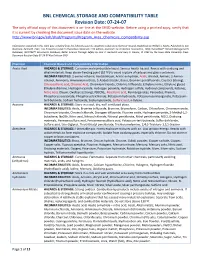
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -
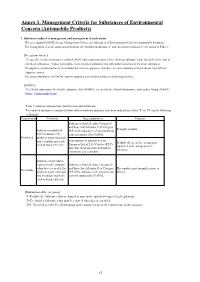
Annex 1. Management Criteria for Substances of Environmental Concern (Automobile Products)
Annex 1. Management Criteria for Substances of Environmental Concern (Automobile Products) 1. Substances subject to management and management classifications We have adopted GADSL for our Management Criteria for Substances of Environmental Concern (Automobile Products). The management classifications and definitions for "prohibited substances" and "declarable substances" are shown in Table 1. [Precautions for use] The specific chemical substances within GADSL only enumerate some of the chemical substances that fall under their class of chemical substances. It does not regulate every chemical substance that falls under said class of chemical substances. The Japanese translation has been included for reference purposes, but there are some substances which do not have official Japanese names. The latest information on GADSL must be obtained and confirmed from the following website. [GADSL] The Global Automotive Declarable Substance List (GADSL) released by the Global Automotive Stakeholder Group (GASG) http://www.gadsl.org/ Table 1. Stanley's management classifications and definitions The reported substances contained within vehicle materials and parts have been indicated as either "P" or "D" via the following definitions. Classification Definition Target substances Response Substances that fall under Category P and those that fall under P of Category Promptly prohibit. Substances prohibited D/P of the substances of environmental from inclusion in the concern stipulated by GADSL Prohibited products, parts, materials, Four substances stipulated in the and secondary materials Prohibit all except for exempt parts European End of Life Vehicles (ELV) used at Stanley Electric stipulated in the European ELV Directive (lead, mercury, hexavalent Directive. chromium, and cadmium) Substances that must be reported to the company Substances that fall under Category D when they are used in the and those that fall under D of Category The supplier must promptly report to Declarable products, parts, materials, D/P of the substances of environmental Stanley. -

Hazardous, Restricted, and Perishable Mail
Hazardous, TransmittalRestricted Letter, and Perishable Mail Publication 52 January 2008 Transmittal Letter A. Purpose. (Edit document ID variable.), Hazardous, Restricted, and Perishable Mail, provides important information to help mailers determine what may be mailed and how certain items must be packaged to keep the mail safe. It also provides guidance to Postal Service™ employees accepting this mail. We publish the standards for hazardous, restricted, and perishable mail in Mailing Standards of the United States Postal Service, Domestic Mail Manual (DMM®) section 601, “Mailability.” The DMM contains all of our official prices and standards for domestic mail, and it governs all other postal information. The DMM is available on Postal Explorer® at pe.usps.com. Customers may also consult their local postmaster or manager of business mail entry for additional information about hazardous materials in the mail. B. Effective Date. This edition is effective January 2008 and updates the online edition dated April 2006. All previous issues of (Edit document ID variable.) are obsolete. C. Availability. Public: The public can access (Edit document ID variable.) in PDF and HTML formats on Postal Explorer at pe.usps.com. Postal Service: Postal Service employees can access (Edit document ID variable.) in PDF and HTML formats on the USPS Web site. D. How to Use This Book. An introduction is located at the beginning of the book. It explains the purpose and scope of the book, as well as the need to protect Postal Service employees and customers from potentially dangerous materials in the mail. In the appendices, we have included a hazardous materials table, packaging instructions for mailable materials, and a glossary of terms. -

Management Standard for Banned & Controlled Substances Uap.01226 Ab
Proprietary Information of: Title: Document No. Rev. MANAGEMENT STANDARD FOR BANNED UAP.01226 AC & CONTROLLED SUBSTANCES For Reference Only When Printed Bez pečiatky len pre informáciu Solo per riferimento quando stampato 打印时仅供参考 Document revision verification may be obtained in Bel Fuse’s PLM system. Record of Revision: Rev Originator Description Approved By Date ECO AA M.Supakova Initial release E. Vergara 2017-04-20 C77008 AB M.Supakova Add limit for Halogen free product in table 1 E. Vergara 2019-10-31 C96739 AC M.Supakova Add TSCA, POPs requirements to table 1, E. Vergara 2021-08-27 CO111522 correct MCV Threshold according to customer specification, add point 6.5 regarding custom and industry specific requirements BPS Document Template UAF.01898 rev AC Page 1 of 112 Proprietary Information of: Title: Document No. Rev. MANAGEMENT STANDARD FOR BANNED UAP.01226 AC & CONTROLLED SUBSTANCES Contents 1 Purpose ...................................................................................................................................................................... 3 2 Scope.......................................................................................................................................................................... 3 3 Reference ................................................................................................................................................................... 3 4 Definition .................................................................................................................................................................... -

149 Subpart B—Table of Hazardous Materials and Special Provisions
Pipeline and Haz. Matls. Safety Admin., DOT § 172.101 Subpart B—Table of Hazardous for describing materials for domestic Materials and Special Provisions transportation but may be inappro- priate for international transportation § 172.101 Purpose and use of haz- under the provisions of international ardous materials table. regulations (e.g., IMO, ICAO). An alter- (a) The Hazardous Materials Table nate proper shipping name may be se- (Table) in this section designates the lected when either domestic or inter- materials listed therein as hazardous national transportation is involved. materials for the purpose of transpor- (4) The letter ‘‘G’’ identifies proper tation of those materials. For each shipping names for which one or more listed material, the Table identifies the technical names of the hazardous ma- hazard class or specifies that the mate- terial must be entered in parentheses, rial is forbidden in transportation, and in association with the basic descrip- gives the proper shipping name or di- tion. (See § 172.203(k).) rects the user to the preferred proper (5) The letter ‘‘I’’ identifies proper shipping name. In addition, the Table shipping names which are appropriate specifies or references requirements in for describing materials in inter- this subchapter pertaining to labeling, national transportation. An alternate packaging, quantity limits aboard air- proper shipping name may be selected craft and stowage of hazardous mate- when only domestic transportation is rials aboard vessels. involved. (b) Column 1: Symbols. Column 1 of the (6) The letter ‘‘W’’ denotes a material Table contains six symbols (‘‘ + ’’, ‘‘A’’, that is subject to the requirements of ‘‘D’’, ‘‘G’’, ‘‘I’’ and ‘‘W’’) as follows: this subchapter only when offered or (1) The plus (+) sign fixes the proper intended for transportation by vessel, shipping name, hazard class and pack- unless the material is a hazardous sub- ing group for that entry without regard stance or a hazardous waste. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0286914 A1 Crowe Et Al
US 20090286914A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0286914 A1 Crowe et al. (43) Pub. Date: Nov. 19, 2009 (54) SEMI-CRYSTALLINE POLYMER Related U.S. Application Data COMPOSITION AND ARTICLE (60) Provisional application No. 60/636,531, filed on Dec. MANUFACTURED THEREFROM 17, 2004, provisional application No. 60/710,161, (75) Inventors: Christie W. Crowe, Alpharetta, GA filed on Aug. 23, 2005. (US); Glenn W. Cupta, Roswell, GA (US) Publication Classification (51) Int. Cl. Correspondence Address: CSK 3/28 (2006.01) OBLON, SPIVAK, MCCLELLAND MAIER & CSK 3/38 (2006.01) NEUSTADT, L.L.P. 194O DUKE STREET (52) U.S. Cl. ......................................... 524/404; 524/428 ALEXANDRIA, VA 22314 (US) (57) ABSTRACT (73) Assignee: SOLVAY ADVANCED Polymer composition comprising (i) at least one semi-crys POLYMERS, L.L.C., Alpharetta, talline polymer (SCP) chosen from polyamides, pol GA (US) yaryletherketones, liquid crystal polymers, polyalkylene Appl. No.: phthalates, polycarbonates, polyarylene Sulphides and pol (21) 11/721,980 yarylene oxides, and (ii) at least one oxide (OX) chosen from (22) PCT Fled: Dec. 15, 2005 acid oxides of an element having an electronegativity of at most 2.2 and amphoteric oxides, and (iii) at least one nitride (86) PCT NO.: PCT/EP2005/056803 (NJ) of an element having an electronegativity of from 1.3 to 2.5. Article comprising at least one part comprising the poly S371 (c)(1), mer composition as above described. Part of an article com (2), (4) Date: Jun. 16, 2007 prising the polymer composition as above described. Patent Application Publication Nov. -

Mercury in Oil Products
Mercury in Oil Products Prologue: It started out as seeing cats “dancing” in the streets, often collapsing and dying...” Minamata Disease a.k.a.: Dancing Cat Disease Discovered in Minamata, Japan in 1956 Caused by methyl mercury in chemical plant wastewater Bioaccumulated in fish & shellfish Seafood eaten by local populace, resulting in mercury poisoning Deaths continued for 36 years Government & company did little to prevent pollution 1,784 deaths, 2,265 victims had been officially recognized, > 10,000 effected Mercury in Crude Oil The United Nations has developed a binding global treaty on the control of mercury release, completed on 19 January 2013 in Geneva, Switzerland - the Minamata Convention. This Treaty is scheduled to be formally adopted and signed at a conference in Minamata, Kumamoto prefecture, Japan from 10 to 11 October 2013. Mercury in Crude Oil UN Committee IPIECA published the largest publicly available dataset on mercury levels covering 446 Crude Oils and Condensates. Mercury in Approximately 3% of crude oils representing less than 1% of Crude Oil global production are above 100 ppb Minamata Convention Discharge , production, export and import of mercury containing products will be banned by 2020. • Batteries, except for 'button cell' batteries used in implantable medical devices • Switches and relays • Certain types of compact fluorescent lamps (CFLs) • Mercury in cold cathode fluorescent lamps and external electrode florescent lamps • Soaps and cosmetics The treaty excludes oil, gas, iron and steel production facilities, allowing parties to determine independently if measures are needed within those categories. Mercury in Crude Oil The Problem In refineries and offshore installations, deposits and small puddles of liquid mercury have been found in crude oil piping and containment. -

UPS Chemical Table
UPS Chemical Table - 49 CFR Version (Ground and Air Packages) BASIC DESCRIPTION FOR GROUND AND AIR GROUND AND AIR HAZARDOUS MATERIALS SHIPMENTS GROUND SHIPMENTS AIR SHIPMENTS SHIPMENTS Symbols: "‡" Requires a Technical Name / "*" For Ltd Qty see 49 CFR Part 173.*** / "**" See 49 CFR 173.27 for inner receptacle requirements / "▲" Accepted as CAO if amount is listed / "♦" PG = "II" or blank / "♦♦" PG = "III" or blank / "#" Sub-risk may be omitted from shipping papers if 49 CFR 172.400a(c) criteria met. (ref 172.202(a)(3)) CARGO HAZARD DOT PASSENGER AIRCRAFT DOT CLASS OR EXEMPTION, GROUND LTD QTY AIRCRAFT MAX NET EXEMPTION, HAZARDOUS MATERIALS DESCRIPTIONS DIVISION I.D. NUMBER LABEL(S) REQUIRED OR SPECIAL SERVICE TO LABEL(S) REQUIRED OR MAX NET MAX NET PER SPECIAL NON-BULK AND PROPER SHIPPING NAME (Subsidiary if (ALSO MARK PACKING EXEMPTION, SPECIAL PERMIT OR PERMIT CANADA EXEMPTION, SPECIAL PERMIT OR PER PER PACKAGE PERMIT SPECIAL EXCEPTIONS PACKAGING (ALSO MARK ON PACKAGE) applicable) ON PACKAGE) GROUP EXCEPTION OR 173.13 PERMITTED EXCEPTION PACKAGE** PACKAGE** ▲ OR 173.13 PROVISIONS §173.*** §173.*** (1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) Accellerene, see p-Nitrosodimethylaniline Accumulators, electric, see Batteries, wet etc Accumulators, pressurized, pneumatic or hydraulic (containing non-flamable gas), see Articles pressurized, pneumatic or hydraulic (containing non-flamable gas) Accumulators, pressurized, pneumatic or hydraulic (containing non-flammable gas), see Articles pressurized, pneumatic or hydraulic -
000090540 DI Stoffliste-Restricted Substances List-Unterschrift
QES Document Stoffliste - Doc No 000090540 AVL LIST GmbH Liste deklarationspflichtiger und verbotener Core process: - Stoffe Revision: 06 Restricted Substances List Gültig ab: 12/2015 Änderungsprotokoll / Revision Protocol Revisionsdatum / Revision Änderungsgrund / Reason of Changes Revision Date 00 01/05/2002 Erstausgabe / first edition 01 15/10/2004 Geänderte Anforderungen / Altered requirements (Ford, Bosch and Volvo) 02 01/02/2008 Geänderte Anforderungen / Altered requirements (Ford, Bosch, Volvo, Renault) 03 10/09/2008 Geänderte Anforderungen / Altered requirements (Ford, Bosch, Volvo, Renault, GADSL) Geänderte Anforderungen / Altered requirements (Ford, Bosch, Volvo, Renault, GADSL, 04 09/03/2009 Scania) 05 20/04/2012 Geänderte Anforderungen / Altered requirements (Bosch, Volvo) Geänderte Anforderungen / Altered requirements (Bosch, Ford, Kubota, Scania, 06 26/11/2015 Volvo) Name Datum / Date Unterschrift / Signature Erstellt / Created O. Herschmann 26/11/2015 Geprüft / Reviewed H. Reitbauer 26/11/2015 Freigegeben / Released O. Herschmann 26/11/2015 Public Copyright © 2015 AVL LIST GMBH, all rights reserved. 000090540_DI_Stoffliste-Restricted Substances List; R06 Uncontrolled print out, controlled version in Intranet. Page 1 of 47 Dok-Nr. 000090540 QES Dokument Kernprozess: - AVL LIST GmbH Stofflisten Revision: 06 Liste deklarationspflichtiger und verbotener Stoffe Gültig ab: 12/2015 Zweck: Dieses QES-Dokument dient der Definition von Stoffen, die nur eingeschränkt oder unter gewissen Rahmenbedingungen in der AVL List GmbH verwendet oder durch sie in Umlauf gebracht werden dürfen. Diese Stoffliste ergänzt die Verantwortung jedes Lieferanten, geltende, möglicherweise schärfere gesetzliche Vorschriften in der jeweils aktuellen Fassung einzuhalten. Geltungsbereich: Dieses QES-Dokument ist für alle Stoffe, Zubereitungen und Produkte inklusive deren Verpackungen anzuwenden, die an AVL geliefert, innerhalb AVL hergestellt, verwendet oder von AVL geliefert werden. -

List/Code of Hazardous Materials
Hazardous Materials Table This Table is a compilation of lists of hazardous materials from the following sources: 1. Environmental Protection Agency (EPA), Title 40 Code of Federal Regulations, Hazardous Waste Regulations 2. Department of Transportation (DOT), Title 49 Code of Federal Regulations, Transportation of Hazardous Materials 3. Califonia Medical Waste Management Act (MWMA) 4. Department of Labor, Occupational Safety & Health Administration, Title 29 Code of Federal Regulations, Subpart Z, Toxic and Hazardous Substances 5. Environmental Protection Agency, Superfund Amendment & Reauthorization Act (SARA)/Title III, Extremely Hazardous Substances 6. American Conference of Governmental Industrial Hygienists, Identification and classification of carcinogens. (1986) Key to Hazard Codes C Corrosive CL Combustible Liquid E Explosive FG Flammable Gas FL Flammable Liquid FS Flammable Solid I Irritating Material NFG Nonflammable Gas OA Otherwise Regulated Material Class A OB Otherwise Regulated Material Class B OC Otherwise Regulated Material Class C OD Otherwise Regulated Material Class D OE Otherwise Regulated Material Class E OG Organic Peroxide OX Oxidizer P Poison R Reactive * Nonhazardous Waste Note: Materials without a hazard code have not been classified and may be hazardous. 1 Hazardous Materials Table N.O.S. Descriptions Acetorphine Acetoxytriphenylstannane Flammable liquid, n.o.s. (FL) Acetyl Bromide (C) Combustible liquid, n.o.s. (CL) Acetyl Chloride (FL, P) Hazardous Waste Liquid, n.o.s. (OE) Acetyl Iodide (C) Acid liquid, n.o.s. (C) Acetylacetone (FL) Flammable liquid, corrosive, n.o.s. (FL, C) 2-Acetylaminofluorene (P) Hazardous Waste Solid, n.o.s. (OE) Acetylbromazine Formaldehyde solution (P, OA) Acetyldihydrocodeine Corrosive solids, n.o.s. (C) Acetylene (FG) Flammable liquid, poisonous, n.o.s.