The Landscape of Extreme Genomic Variation in the Highly Adaptable Atlantic Killifish
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deterministic Shifts in Molecular Evolution Correlate with Convergence to Annualism in Killifishes
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.09.455723; this version posted August 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Deterministic shifts in molecular evolution correlate with convergence to annualism in killifishes Andrew W. Thompson1,2, Amanda C. Black3, Yu Huang4,5,6 Qiong Shi4,5 Andrew I. Furness7, Ingo, Braasch1,2, Federico G. Hoffmann3, and Guillermo Ortí6 1Department of Integrative Biology, Michigan State University, East Lansing, Michigan 48823, USA. 2Ecology, Evolution & Behavior Program, Michigan State University, East Lansing, MI, USA. 3Department of Biochemistry, Molecular Biology, Entomology, & Plant Pathology, Mississippi State University, Starkville, MS 39759, USA. 4Shenzhen Key Lab of Marine Genomics, Guangdong Provincial Key Lab of Molecular Breeding in Marine Economic Animals, BGI Marine, Shenzhen 518083, China. 5BGI Education Center, University of Chinese Academy of Sciences, Shenzhen 518083, China. 6Department of Biological Sciences, The George Washington University, Washington, DC 20052, USA. 7Department of Biological and Marine Sciences, University of Hull, UK. Corresponding author: Andrew W. Thompson, [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.08.09.455723; this version posted August 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract: The repeated evolution of novel life histories correlating with ecological variables offer opportunities to test scenarios of convergence and determinism in genetic, developmental, and metabolic features. Here we leverage the diversity of aplocheiloid killifishes, a clade of teleost fishes that contains over 750 species on three continents. -

The Mystery of the Banded Killifish Fundulus Diaphanus Population Explosion: Where Did They All Come From?
The Mystery of the Banded KillifishFundulus ( diaphanus) Population Explosion: Where Did They All Come from? Philip W. Willink, Jeremy S. Tiemann, Joshua L. Sherwood, Eric R. Larson, Abe Otten, Brian Zimmerman 3 American Currents Vol. 44, No. 4 THE MYSTERY OF THE BANDED KILLIFISH FUNDULUS DIAPHANUS POPULATION EXPLOSION: WHERE DID THEY ALL COME FROM? Philip W. Willink, Jeremy S. Tiemann, Joshua L. Sherwood, La Grange Park, IL Illinois Natural History Survey Illinois Natural History Survey Eric R. Larson, Abe Otten, Brian Zimmerman University of Illinois at Scott Community The Ohio State University Urbana-Champaign College Stream and River Ecology Lab Banded Killifish Fundulus diaphanus are no strangers to NAN- and have not been seen since, they were only known from a hand- FAers. Over the past several years, there have been multiple arti- ful of inland lakes in the far northeastern corner the state (Fig. 1). cles in American Currents covering their distribution (Hatch 2015; Even there, population numbers were low. Schmidt 2016a, 2018; Olson and Schmidt 2018; Li 2019), stocking So it was with great excitement that in the early 2000s Illinois to restore populations (Bland 2013; Schmidt 2014), and appear- ichthyologists started to find more and more presumed Western ance in a hatchery (Schmidt 2016b). Their range extends from the Banded Killifish in Lake Michigan (Willink et al. 2018). They Canadian Maritime provinces south along the Atlantic coast to were even showing up in downtown Chicago (Willink 2011). It the Carolinas, as well as westward through the Great Lakes region was hoped that this range expansion was evidence of an uncom- to the upper Mississippi watershed. -

Gene Expression After Freshwater Transfer in Gills and Opercular Epithelia of Killifish: Insight Into Divergent Mechanisms of Ion Transport Graham R
The Journal of Experimental Biology 208, 2719-2729 2719 Published by The Company of Biologists 2005 doi:10.1242/jeb.01688 Gene expression after freshwater transfer in gills and opercular epithelia of killifish: insight into divergent mechanisms of ion transport Graham R. Scott1,*, James B. Claiborne2, Susan L. Edwards2,3, Patricia M. Schulte1 and Chris M. Wood4 1Department of Zoology, University of British Columbia, Vancouver BC, Canada V6T 1Z4, 2Department of Biology, Georgia Southern University, Statesboro, GA 30460-8042, USA, 3Department of Physiology and Pharmacology, James Cook University, Cairns, QLD 4879, Australia and 4Department of Biology, McMaster University, Hamilton ON, Canada L8S 4K1 *Author for correspondence (e-mail: [email protected]) Accepted 17 May 2005 Summary We have explored the molecular basis for differences in epithelium, NHE2 was not expressed; furthermore, physiological function between the gills and opercular Na+,K+-ATPase activity was unchanged after transfer to epithelium of the euryhaline killifish Fundulus freshwater, CA2 mRNA levels decreased, and NHE3 levels heteroclitus. These tissues are functionally similar in increased. Consistent with their functional similarities in seawater, but in freshwater the gills actively absorb Na+ seawater, killifish gills and opercular epithelium expressed – + + + + – but not Cl , whereas the opercular epithelium actively Na ,K -ATPase α1a, Na ,K ,2Cl cotransporter 1 absorbs Cl– but not Na+. These differences in freshwater (NKCC1), cystic fibrosis transmembrane conductance physiology are likely due to differences in absolute levels regulator (CFTR) Cl– channel and the signalling protein of gene expression (measured using real-time PCR), as 14-3-3a at similar absolute levels. Furthermore, NKCC1 several proteins important for Na+ transport, namely and CFTR were suppressed equally in each tissue after Na+,H+-exchanger 2 (NHE2), carbonic anhydrase 2 (CA2), freshwater transfer, and 14-3-3a mRNA increased in both. -
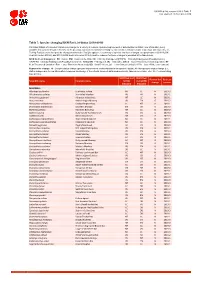
Table 7: Species Changing IUCN Red List Status (2018-2019)
IUCN Red List version 2019-3: Table 7 Last Updated: 10 December 2019 Table 7: Species changing IUCN Red List Status (2018-2019) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2018 (IUCN Red List version 2018-2) and 2019 (IUCN Red List version 2019-3) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered [CR(PE) - Critically Endangered (Possibly Extinct), CR(PEW) - Critically Endangered (Possibly Extinct in the Wild)], EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2018) List (2019) change version Category -

The African Turquoise Killifish: a Model for Exploring Vertebrate Aging and Diseases in the Fast Lane
Downloaded from symposium.cshlp.org on June 11, 2016 - Published by Cold Spring Harbor Laboratory Press The African Turquoise Killifish: A Model for Exploring Vertebrate Aging and Diseases in the Fast Lane 1 1,2 ITAMAR HAREL AND ANNE BRUNET 1Department of Genetics, Stanford University, Stanford, California 94305 2Glenn Laboratories for the Biology of Aging at Stanford, Stanford, California 94305 Correspondence: [email protected] Why and how organisms age remains a mystery, and it defines one of the biggest challenges in biology. Aging is also the primary risk factor for many human pathologies, such as cancer, diabetes, cardiovascular diseases, and neurodegenerative diseases. Thus, manipulating the aging rate and potentially postponing the onset of these devastating diseases could have a tremendous impact on human health. Recent studies, relying primarily on nonvertebrate short-lived model systems, have shown the importance of both genetic and environmental factors in modulating the aging rate. However, relatively little is known about aging in vertebrates or what processes may be unique and specific to these complex organisms. Here we discuss how advances in genomics and genome editing have significantly expanded our ability to probe the aging process in a vertebrate system. We highlight recent findings from a naturally short-lived vertebrate, the African turquoise killifish, which provides an attractive platform for exploring mechanisms underlying vertebrate aging and age-related diseases. MODELING AGING IN THE ing vertebrate aging and age-related diseases (Harel et al. LABORATORY 2015). Aging research has greatly benefited from investigating short-lived nonvertebrate models (Fig. 1A), specifically THE AFRICAN TURQUOISE KILLIFISH yeast (Saccharomyces cerevisiae), worm (Caenorhabdi- tis elegans), and fly (Drosophila melanogaster). -

Draft Risk Assessment Report Nothobranchius Furzeri (Turquoise
APPLICATION TO AMEND THE LIST OF SPECIMENS SUITABLE FOR LIVE IMPORT Draft Risk Assessment Report 1. Taxonomy of the species a. Family name: Aplocheilidae b. Genus name: Nothobranchius c. Species: N. furzeri d. Subspecies: No e. Taxonomic Reference: http://www.uniprot.org/taxonomy/105023 f. Common Names: Turquoise killifish g. Is the species a genetically-modified organism (GMO)? No 2. Status of the species under CITES Species is not listed in the CITES Appendices. 3. Ecology of the species a. Longevity: what is the average lifespan of the species in the wild and in captivity? The average lifespan of the species in captivity is 13 weeks (1). Wild derived N. furzeri from three different habitats showed a maximum lifespan of 25-32 weeks (1). b. What is the maximum length and weight that the species attains? Provide information on the size and weight range for males and females of the species. As per the original formal description of the species, the standard length of N. furzeri from a wild population ranged from 2.5cm to 4.4cm (2). The maximum length the species can attain is 7cm (3). The mean maximal body weight reported for males is 3.8 ± 1.1 g (range: 0.5 – 6.7 g) and 2.2 ± 0.6 g (range: 0.2 - 3.6 g) for females (4). Growth rates of Nothobranchius in the wild have been reported to be similar to those in captivity (5). c. Discuss the identification of the individuals in this species, including if the sexes of the species are readily distinguishable, and if the species is difficult to distinguish from other species. -

Gill Trematodes (Flukes) in Wild-Caught Killifish (<I>Fundulus
NOTES Gill Trematodes (Flukes) in Wild-Caught Killifish (Fundulus heteroclitus) DAVID R. GOULDING, BS, RLAT,1* TERRY L. BLANKENSHIP-PARIS, DVM, MS, DIPLOMATE, ACLAM,1 GREGORY A. LEWBART, MS, VMD, DIPLOMATE, ACZM,2 PAGE H. MYERS, BS, LATG,1 TRACY K. DEMIANENKO, BS, RLAT,1 JAMES A. CLARK, LATG,1 AND DIANE B. FORSYTHE, DVM, DIPLOMATE, ACLAM1 Three wild caught killifish (Fundulus heteroclitus) on an Institutional Animal Care and Use Committee-approved protocol were found dead within 2 days after being received. The fish were housed in two separate aquaria. Aquarium water was evaluated, and pH, salinity, ammonia, nitrate, and nitrite levels were within acceptable parameters. Several remaining fish appeared to be slow-moving and were presented for necropsy. Multiple, scattered, ulcerated skin lesions (diameter, 1 to 5 mm) were noted at necropsy and were cultured. No pathogenic bacteria were isolated. Wet-mount samples of the gills revealed multiple cysts at the gill margins, each containing a motile organism. No other gill parasites were detected. A diagnosis of trematodiasis was made. The cysts were identified as encysted metacercariae of a digenetic trematode. We surmise that the large numbers of gill flukes combined with the stress of recent shipment likely caused the observed morbidity and mortality. Sixty-two wild-caught killifish (Fundulus heteroclitus) obtained from Duke Marine Center (Beaufort, N.C.) were group-housed upon receipt in two 38-gallon aquaria containing conditioned brack- ish water. Thirty-six fish were placed in tank A, and 26 fish were placed in tank B. One day after arrival, two fish were found dead in tank B, and 2 days after arrival, one fish was found dead in tank A. -

Compare and Contrast the Water Environment Between Death Valley Pupfish Specie and Devil’S Hole Pupfish Specie
Compare and Contrast the Water environment between Death Valley Pupfish Specie and Devil’s Hole Pupfish Specie By Roy Tianran Gao 1 Table of Contents Title page 1 Abstract 3 Introduction and Background 3 Water Temperature 4 Salinity 6 Water Level 7 Conservation 10 Conclusion 11 References 12 2 ABSTRACT The two types of pupfish (Cyprinodon) in Death Valley National Park are Death Valley pupfish and Devil’s Hole pupfish. Death Valley pupfish has been existed over 10,000 years and Devil’s Hole pupfish has been existed for over 20,000 years. Both of the pupfishes are endangered species. The average number of Death Valley pupfish has decreased by about 100 since 1990s, and the number of Devil’s Hole pupfish has decreased by 400 since 1995. Comparing the water level, water temperature and the water salinity between the two species of pupfish would help to define the living requirements and reason of decreasing population. The research toward the result is based on 7 journal articles, 4 websites, and 1 book. As the result shows, Death Valley Pupfish and Devil’s Hole Pupfish live in different water environments and functioned differently. Understanding the water environment of the two types of pupfishes will help people building new habitats for pupfishes and increase their population so that would be possible to avoid the extinction of pupfishes from the earth. INTRODUCTION AND BACKGROUND Pupfish is a small killifish in the Southwest of America. There are five pupfish species in Death Valley which are Armargosa pupfish, Saratoga Pupfish, Devil’s Hole pupfish, Death Valley pupfish, and Cotton ball Marsh pupfish (National Park Service, 2008). -

Supplemental Methods
Supplemental Methods Collection and Preparation After extraction, the DNA samples were sheared with a 1.5 blunt end needles (Jensen Global, Santa Barbara, CA, USA) and run through pulse field gel electrophoresis, in order to separate large DNA molecules, for 16 hours. Samples were checked with a spectrophotometer (Synergy H1 Hybrid Reader, BioTek Instruments Inc., Winooski, VT) and Qubit® fluorometer for quantification of our genomic DNA. Illumina and Pacbio Hybrid Assembly, Genome Size Estimation, and Quality Assessment All computational and bioinformatics analyses were conducted on the High Performance Computing (HPC) Cluster located that University of California, Irvine. Sequence data generated from two lanes of Illumina HiSeq 2500 were concatenated and raw sequence reads were assembled through PLATANUS v1.2.1 (1), which accounts for heterozygous diploid sequence data. Parameters used for PLATANUS was -m 256 (memory) and -t 48 (threads) for this initial assembly. Afterwards, contigs assembled from PLATANUS and reads from 40 SMRT cells of PacBio sequencing were assembled with a hybrid assembler DBG2OLC v1.0 (2). We used the following parameters in DBG2OLC: k 17 KmerCovTh 2 MinOverlap 20 AdaptiveTh 0.01 LD1 0 and RemoveChimera 1 and ran pbdagcon with default parameters. Without Illumina sequence reads, we also conducted a PacBio reads only assembly with FALCON v0.3.0 (https://github.com/PacificBiosciences/FALCON) with default parameters in order to assemble PacBio reads into contiguous sequences. The parameters we used were input type = raw, length_cutoff = 4000, length_cutoff_pr = 8000, with different cluster settings 32, 16, 32, 8, 64, and 32 cores, concurrency setting jobs were 32, and the remaining were default parameters. -

Cyprinodontiformes, Nothobranchiidae)
COMPARATIVE A peer-reviewed open-access journal CompCytogen 10(3): Divergent439–445 (2016) karyotypes of the annual killifish genusNothobranchius ... 439 doi: 10.3897/CompCytogen.v10i3.9863 SHORT COMMUNICATION Cytogenetics http://compcytogen.pensoft.net International Journal of Plant & Animal Cytogenetics, Karyosystematics, and Molecular Systematics Divergent karyotypes of the annual killifish genus Nothobranchius (Cyprinodontiformes, Nothobranchiidae) Eugene Krysanov1, Tatiana Demidova1, Bela Nagy2 1 Severtsov Institute of Ecology and Evolution, Russian Academy of Sciences, Leninsky prospect, Moscow, 119071 Russia 2 30, rue du Mont Ussy, 77300 Fontainebleau, France Corresponding author: Eugene Krysanov ([email protected]) Academic editor: I. Kuznetsova | Received 13 July 2016 | Accepted 18 August 2016 | Published 16 September 2016 http://zoobank.org/516FBC12-825D-46E0-BAAA-25ABA00F8607 Citation: Krysanov E, Demidova T, Nagy B (2016) Divergent karyotypes of the annual killifish genus Nothobranchius (Cyprinodontiformes; Nothobranchiidae). Comparative Cytogenetics 10(3): 439–445. doi: 10.3897/CompCytogen. v10i3.9863 Abstract Karyotypes of two species of the African annual killifish genus Nothobranchius Peters, 1868, N. brieni Poll, 1938 and Nothobranchius sp. from Kasenga (D.R. Congo) are described. Both species displayed dip- loid chromosome number 2n = 49/50 for males and females respectively with multiple-sex chromosome system type X1X2Y/X1X1X2X2. The karyotypes of studied species are considerably different from those previously reported for the genus Nothobranchius and similar to the Actinopterygii conservative karyotype. Keywords Africa, chromosome number, karyotype, killifish, Nothobranchius Introduction Annual killifishes belonging to the genusNothobranchius Peters, 1868 are mainly dis- tributed in eastern Africa but several species are found in central Africa (Wildekamp 2004). They inhabit temporary pools that dry out during the dry season and have spe- cific adaptations for extreme environments. -

Recovery Plan Endangered and Species Nevada
RECOVERYPLAN FOR THE ENDANGEREDAND THREATENED SPECIES OF&H MEADOWS, NEVADA Prepared by Don W. Sada U.S. Fish and Wildlife Service Reno, Nevada RECOVERY PLAN FOR THE ENDANGERED AND THREATENED SPECIES OF ASH MEADOWS, NEVADA Prepared By Don W. Sada U.S. Fish and Wildlife Service Reno, Nevada for the U.S. Fish and Wildlife Service Portland, Oregon ~FP2 3 ‘:XN Date This plan covers the following federally listed species in Ash Meadows, Nevada and California: Devil’s Hole pupfish, Warm Springs pupfish, Ash Meadows Arnargosa pupfish, Ash Meadows speckled dace, Ash Meadows naucorid, Ash Meadows blazing star, Ash Meadows ivesia, Ainargosa niterwort, Spring-loving centaury, Ash Meadows sunray, Ash Meadows inilk-vetch, and Ash Meadows guxnplant. THIS IS THE COMPLETED ASH MEADOWS SPECIES RECOVERY PLAN. IT HAS BEEN APPROVED BY THE U.S. FISH AND WILDLIFE SERVICE. IT DOES NOT NECESSARILY REPRESENT OFFICIAL POSITIONS OR APPROVALS OF COOPERATING AGENCIES (AND IT DOES NOT NECESSARILY REPRESENT THE VIEWS OF ALL INDIVIDUALS) WHO PLAYED THE KEY ROLE IN PREPARING THIS PLAN. THIS PLAN IS SUBJECT TO MODIFICATION AS DICTATED BY NEW FINDINGS AND CHANGES IN SPECIES STATUS, AND COMPLETION OF TASKS DESCRIBED IN THE PLAN. GOALS AND OBJECTIVES WILL BE ATTAINED AND FUNDS EXPENDED CONTINGENT UPON APPROPRIATIONS, PRIORITIES, AND OTHER BUDGETARY CONSTRAINTS. LITERATURE CITATION SHOULD READ AS FOLLOWS U.S. Fish and Wildlife Service. 1990. Recovery plan for the endangered and threatened species of Ash Meadows, Nevada. U.S. Fish and Wildlife Service, Portland, Oregon. 123 pp. Additional copies may be obtained from Fish and Wildlife Reference Service 5430 Grosvenor Lane, Suite 110 Bethesda, Maryland 20814 Telephone: 301-492-6403 1-800-582-3421 : ACKNOWLEDGMENTS: This plan results from the efforts of many who spent considerable time and energy to prevent the destruction of Ash Meadows and the extinction of its diverse endemic biota. -

Evolutionary History and Whole Genome Sequence of Pejerrey (Odontesthes Bonariensis): New Insights Into Sex Determination in Fishes
Evolutionary History and Whole Genome Sequence of Pejerrey (Odontesthes bonariensis): New Insights into Sex Determination in Fishes by Daniela Campanella B.Sc. in Biology, July 2009, Universidad Nacional de La Plata, Argentina A Dissertation submitted to The Faculty of The Columbian College of Arts and Sciences of The George Washington University in partial fulfillment of the requirements for the degree of Doctor of Philosophy January 31, 2015 Dissertation co-directed by Guillermo Ortí Louis Weintraub Professor of Biology Elisabet Caler Program Director at National Heart, Lung and Blood Institute, NIH The Columbian College of Arts and Sciences of The George Washington University certifies that Daniela Campanella has passed the Final Examination for the degree of Doctor of Philosophy as of December 12th, 2014. This is the final and approved form of the dissertation. Evolutionary History and Whole Genome Sequence of Pejerrey (Odontesthes bonariensis): New Insights into Sex Determination in Fishes Daniela Campanella Dissertation Research Committee: Guillermo Ortí, Louis Weintraub Professor of Biology, Dissertation Co-Director Elisabet Caler, Program Director at National Heart, Lung and Blood Institute, NIH, Dissertation Co-Director Hernán Lorenzi, Assistant Professor in Bioinformatics Department, J. Craig Venter Institute Rockville Maryland, Committee Member Jeremy Goecks, Assistant Professor of Computational Biology, Committee Member ! ""! ! Copyright 2015 by Daniela Campanella All rights reserved ! """! Dedication The author wishes to dedicate this dissertation to: My love, Ford, for his unconditional support and inspiration. For teaching me that admiration towards each other’s work is the fundamental fuel to go anywhere. My family and friends, for being there, meaning “there” everywhere and whenever. My grandpa Hugo, a pejerrey lover who knew how to fish, cook and enjoy the “silver arrows”.