Deterministic Shifts in Molecular Evolution Correlate with Convergence to Annualism in Killifishes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

§4-71-6.5 LIST of CONDITIONALLY APPROVED ANIMALS November
§4-71-6.5 LIST OF CONDITIONALLY APPROVED ANIMALS November 28, 2006 SCIENTIFIC NAME COMMON NAME INVERTEBRATES PHYLUM Annelida CLASS Oligochaeta ORDER Plesiopora FAMILY Tubificidae Tubifex (all species in genus) worm, tubifex PHYLUM Arthropoda CLASS Crustacea ORDER Anostraca FAMILY Artemiidae Artemia (all species in genus) shrimp, brine ORDER Cladocera FAMILY Daphnidae Daphnia (all species in genus) flea, water ORDER Decapoda FAMILY Atelecyclidae Erimacrus isenbeckii crab, horsehair FAMILY Cancridae Cancer antennarius crab, California rock Cancer anthonyi crab, yellowstone Cancer borealis crab, Jonah Cancer magister crab, dungeness Cancer productus crab, rock (red) FAMILY Geryonidae Geryon affinis crab, golden FAMILY Lithodidae Paralithodes camtschatica crab, Alaskan king FAMILY Majidae Chionocetes bairdi crab, snow Chionocetes opilio crab, snow 1 CONDITIONAL ANIMAL LIST §4-71-6.5 SCIENTIFIC NAME COMMON NAME Chionocetes tanneri crab, snow FAMILY Nephropidae Homarus (all species in genus) lobster, true FAMILY Palaemonidae Macrobrachium lar shrimp, freshwater Macrobrachium rosenbergi prawn, giant long-legged FAMILY Palinuridae Jasus (all species in genus) crayfish, saltwater; lobster Panulirus argus lobster, Atlantic spiny Panulirus longipes femoristriga crayfish, saltwater Panulirus pencillatus lobster, spiny FAMILY Portunidae Callinectes sapidus crab, blue Scylla serrata crab, Samoan; serrate, swimming FAMILY Raninidae Ranina ranina crab, spanner; red frog, Hawaiian CLASS Insecta ORDER Coleoptera FAMILY Tenebrionidae Tenebrio molitor mealworm, -

Discovery of the Critically Endangered Annual Killifish, Austrolebias
JoTT SHORT COMMUNI C ATION 2(11): 1282-1285 Discovery of the Critically Endangered annual killifishAustrolebias wolterstorffi (Ahl, 1924) (Cyprinodontiformes: Rivulidae) in Lagoa do Peixe National Park, Rio Grande do Sul, southern Brazil L.E.K. Lanés 1 & L. Maltchik 2 1,2 Laboratory of Ecology and Conservation of Aquatic Ecosystems, University of Vale do Rio dos Sinos (UNISINOS). Av. Unisinos 950, Bairro Cristo Rei, CEP 93022-000, São Leopoldo, Rio Grande do Sul, Brazil. Email: 1 [email protected], 2 [email protected] Abstract: This paper documents the discovery of the rare and live in shallow temporary wetlands formed close to rivers, Critically Endangered annual killifish Austrolebias wolterstorffi streams and lagoons during the rainy season, and die in temporary wetland of Lagoa do Peixe National Park, an im- portant conservation unit of southern Brazil protected under the off when the pools dry (Costa 2003, 2006). Their eggs Ramsar Convention and recognized Biosphere Reserve. survive the dry season and hatch after the next rainy season begins. Keywords: Annual fish, Cynolebiasinae, endemic, freshwater, The genus Austrolebias has recently been redefined Neotropics, protected areas, Ramsar sites, restricted-range fish- phylogenetically by Costa (2006), including the species es, threatened species, wetlands. previously referred to the genus Megalebias Costa. Subsequently the genus Austrolebias was divided into subgenera (Costa 2008), according to clades defined in The neotropical aplocheiloid genus Austrolebias Costa (2006). Accordingly the subgenus Megalebias Costa comprises 40 small annual killifishes endemic to which has the same general geographic range as South America, distributed in southern Brazil, southern Austrolebias, comprises five valid species: Austrolebias Bolivia, Paraguay, Uruguay and northeastern Argentina cheradophilus (Vaz-Ferreira, Sierra de Soriano & Scaglia- (Costa 2010). -

A New Genus of Miniature Cynolebiasine from the Atlantic
64 (1): 23 – 33 © Senckenberg Gesellschaft für Naturforschung, 2014. 16.5.2014 A new genus of miniature cynolebiasine from the Atlantic Forest and alternative biogeographical explanations for seasonal killifish distribution patterns in South America (Cyprinodontiformes: Rivulidae) Wilson J. E. M. Costa Laboratório de Sistemática e Evolução de Peixes Teleósteos, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Caixa Postal 68049, CEP 21944 – 970, Rio de Janeiro, Brasil; wcosta(at)acd.ufrj.br Accepted 21.ii.2014. Published online at www.senckenberg.de/vertebrate-zoology on 30.iv.2014. Abstract The analysis of 78 morphological characters for 16 species representing all the lineages of the tribe Cynopoecilini and three out-groups, indicates that the incertae sedis miniature species ‘Leptolebias’ leitaoi Cruz & Peixoto is the sister group of a clade comprising the genera Leptolebias, Campellolebias, and Cynopoecilus, consequently recognised as the only member of a new genus. Mucurilebias gen. nov. is diagnosed by seven autapomorphies: eye occupying great part of head side, low number of caudal-fin rays (21), distal portion of epural much broader than distal portion of parhypural, an oblique red bar through opercle in both sexes, isthmus bright red in males, a white stripe on the distal margin of the dorsal fin in males, and a red stripe on the distal margin of the anal fin in males.Mucurilebias leitaoi is an endangered seasonal species endemic to the Mucuri river basin. The biogeographical analysis of genera of the subfamily Cynolebiasinae using a dispersal-vicariance, event-based parsimony approach indicates that distribution of South American killifishes may be broadly shaped by dispersal events. -

Laimosemion Ubim, a New Miniature Killifish from the Brazilian Amazon (Teleostei: Rivulidae)
371 Ichthyol. Explor. Freshwaters, Vol. 24, No. 4, pp. 371-378, 3 figs., 1 tab., April 2014 © 2014 by Verlag Dr. Friedrich Pfeil, München, Germany – ISSN 0936-9902 Laimosemion ubim, a new miniature killifish from the Brazilian Amazon (Teleostei: Rivulidae) Wilson J. E. M. Costa* and Henrique Lazzarotto** Laimosemion ubim, new species, is described from a small stream tributary of Lago Amanã system, Central Ama- zon, northern Brazil, based on external and internal anatomical morphological characters. It is considered closely related to other species of Laimosemion, subgenus Owiyeye, from the same region. It is distinguished from all other rivulids by having double-branched epipleural ribs, a condition never found among cyprinodontiforms, and from all its congeners by having hypertrophied teeth on the anterior portion of the outer row of the premax- illa and dentary in males. It reaches a maximum adult size of about 18 mm SL and exhibits several reductive characters, as expected for a miniature species, including a notable reductive character state – four branchiostegal rays. Introduction new reports about miniature freshwater fishes (i. e., maturing at sizes under 20 mm SL, or reach- Evolutionary events of miniaturization, consisting ing 26.0 mm SL as maximum adult size, as estab- of extreme reduction in body size within a lineage lished by Weitzman & Vari (1988), have became (Hanken & Wake, 1993), are a common phenom- frequent in the literature (Mattos et al., 2013). enon among freshwater teleost fishes in tropical A great part of reported miniaturization events regions of the world (e. g., Weitzman & Vari, 1988; in freshwater fishes involves species inhabiting Kottelat & Vidthayanon, 1993; Costa & Le Bail, swampy forests with acidic waters (e. -

Unrecognized Biodiversity in a World's Hotspot: Three New Species Of
Creative Commons Attribution 4.0 licence (CC-BY); original download https://pensoft.net/journals Zoosyst. Evol. 94 (2) 2018, 263–280 | DOI 10.3897/zse.94.24406 Unrecognized biodiversity in a world’s hotspot: three new species of Melanorivulus (Cyprinodontiformes: Rivulidae) from tributaries of the right bank of the Rio Paraná basin, Brazilian Cerrado Matheus V. Volcan1, Francisco Severo-Neto2,3, Luis Esteban K. Lanés1,4 1 Instituto Pró-Pampa (IPPampa), Laboratório de Ictiologia. Rua Uruguay, 1242, Bairro Centro, CEP 96010-630, Pelotas, Rio Grande do Sul, Brasil 2 Universidade Federal de Mato Grosso do Sul, Centro de Biociências, Cidade Universitária, CEP 79070-700, Campo Grande, MS, Brasil 3 Instituto de Biociências, Letras e Ciências Exatas, Departamento de Zoologia e Botânica, Laboratório de Ictiologia, Universidade Estadual Paulista “Júlio de Mesquita Filho” (UNESP), Rua Cristóvão Colombo, 2265, Jardim Nazareth, CEP15054-000, São José do Rio Preto, SP, Brasil 4 Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS), Programa de Pós-Graduação em Zoologia, Laboratório de Fisiologia da Conservação. Avenida Ipiranga, 6681 Pd.12, Bloco C, Sala 250 CP. 1429, CEP 90619–900, Porto Alegre, RS, Brasil http://zoobank.org/BCA6F985-BE53-47C5-B64D-005DA0DD69BE Corresponding author: Matheus V. Volcan ([email protected]) Abstract Received 12 February 2018 The genus Melanorivulus presents a wide geographical distribution in the Neotropical re- Accepted 6 April 2018 gion. Among Melanorivulus, the M. pictus species group has currently 18 species distrib- Published 18 April 2018 uted in the Brazilian Cerrado, mainly in the upper Rio Paraná basin, with some species occurrences in the upper Rio Araguaia and Tocantins. -

Cynolebias (Pisces: Cyprinodontiformes, Rivulidae) Using Banding Techniques
Analysis of Karyotypic Evolution in Natural Populations of Cynolebias (Pisces: Cyprinodontiformes, Rivulidae) Using Banding Techniques G. Garcia1, E. Scvortzoff1, M. C. Maspoli2 and R. Vaz-Ferreira2 1Instituto de Biologia, Departamento de Genetica , Facultad de Ciencias, Tristan Narvaja 1674. 11200 Montevideo, Uruguay. 2Instituto de Biologia , Departamento de Zoologia, Facultad de Ciencias, Tristan Narvaja 1674. 11200 Montevideo, Uruguay. Accepted November 13, 1992 Many genera belonging to the family Rivulidae (Pisces, Cyprinodontiformes) show _??_ evolutionary tendency to a decrease in their chromosome numbers, considering that the ba_??_ chromosomal number found in Teleosts is 2n=48. This process is accompanied by an _??_ crease in the number of biarmed chromosomes. Studying the karyotypes of 127 genera _??_ Rivulinae, Scheel (1972) postulated two types of chromosomal rearrangements alternating _??_ the evolution of these karyotypes: pericentric inversions and centric fusions. This autl suggested that centric fusions can occur between either subtelocentric or acrocentric chron _??_ somes (ST-A), giving rise to metacentrics. Pericentric inversions would then transform th_??_ metacentric chromosomes into larger telocentrics. These pericentric inversions would t_??_ be followed by new centric fusions, thus generating especially large biarmed chromosom_??_ Under this apparently simple reorganization system, parallel evolution would have occur_??_ in many genera. Scheel (op. cit.) particularly described the chromosomal differences found _??_ species of the genus Aphyosemion. The genus Cynolebias Steindachner 1876 is considered to be biologically related to _??_ genera Aphyosemion and Notobranchius because all their species have annual life cycles. _??_ species of Cynolebias originally proved to be chromosomally uniform, having 48 acrocen_??_ chromosomes (Sofa et al. -

The Mystery of the Banded Killifish Fundulus Diaphanus Population Explosion: Where Did They All Come From?
The Mystery of the Banded KillifishFundulus ( diaphanus) Population Explosion: Where Did They All Come from? Philip W. Willink, Jeremy S. Tiemann, Joshua L. Sherwood, Eric R. Larson, Abe Otten, Brian Zimmerman 3 American Currents Vol. 44, No. 4 THE MYSTERY OF THE BANDED KILLIFISH FUNDULUS DIAPHANUS POPULATION EXPLOSION: WHERE DID THEY ALL COME FROM? Philip W. Willink, Jeremy S. Tiemann, Joshua L. Sherwood, La Grange Park, IL Illinois Natural History Survey Illinois Natural History Survey Eric R. Larson, Abe Otten, Brian Zimmerman University of Illinois at Scott Community The Ohio State University Urbana-Champaign College Stream and River Ecology Lab Banded Killifish Fundulus diaphanus are no strangers to NAN- and have not been seen since, they were only known from a hand- FAers. Over the past several years, there have been multiple arti- ful of inland lakes in the far northeastern corner the state (Fig. 1). cles in American Currents covering their distribution (Hatch 2015; Even there, population numbers were low. Schmidt 2016a, 2018; Olson and Schmidt 2018; Li 2019), stocking So it was with great excitement that in the early 2000s Illinois to restore populations (Bland 2013; Schmidt 2014), and appear- ichthyologists started to find more and more presumed Western ance in a hatchery (Schmidt 2016b). Their range extends from the Banded Killifish in Lake Michigan (Willink et al. 2018). They Canadian Maritime provinces south along the Atlantic coast to were even showing up in downtown Chicago (Willink 2011). It the Carolinas, as well as westward through the Great Lakes region was hoped that this range expansion was evidence of an uncom- to the upper Mississippi watershed. -

Characterization of the G Protein-Coupled Receptor Family
www.nature.com/scientificreports OPEN Characterization of the G protein‑coupled receptor family SREB across fsh evolution Timothy S. Breton1*, William G. B. Sampson1, Benjamin Cliford2, Anyssa M. Phaneuf1, Ilze Smidt3, Tamera True1, Andrew R. Wilcox1, Taylor Lipscomb4,5, Casey Murray4 & Matthew A. DiMaggio4 The SREB (Super‑conserved Receptors Expressed in Brain) family of G protein‑coupled receptors is highly conserved across vertebrates and consists of three members: SREB1 (orphan receptor GPR27), SREB2 (GPR85), and SREB3 (GPR173). Ligands for these receptors are largely unknown or only recently identifed, and functions for all three are still beginning to be understood, including roles in glucose homeostasis, neurogenesis, and hypothalamic control of reproduction. In addition to the brain, all three are expressed in gonads, but relatively few studies have focused on this, especially in non‑mammalian models or in an integrated approach across the entire receptor family. The purpose of this study was to more fully characterize sreb genes in fsh, using comparative genomics and gonadal expression analyses in fve diverse ray‑fnned (Actinopterygii) species across evolution. Several unique characteristics were identifed in fsh, including: (1) a novel, fourth euteleost‑specifc gene (sreb3b or gpr173b) that likely emerged from a copy of sreb3 in a separate event after the teleost whole genome duplication, (2) sreb3a gene loss in Order Cyprinodontiformes, and (3) expression diferences between a gar species and teleosts. Overall, gonadal patterns suggested an important role for all sreb genes in teleost testicular development, while gar were characterized by greater ovarian expression that may refect similar roles to mammals. The novel sreb3b gene was also characterized by several unique features, including divergent but highly conserved amino acid positions, and elevated brain expression in pufer (Dichotomyctere nigroviridis) that more closely matched sreb2, not sreb3a. -

2010 by Lee Harper, 2011-2018 Compiled by R. Mccabe .Xls
JAKA INDEX 1962- 2010 by Lee Harper, 2011-2018 compiled by R. McCabe .xls First Last Document Volume Issue Year Date Title Author Page Page Killie Notes 1 1 1962 3 4 February-62 A Chartered Flight Albert J. Klee Killie Notes 1 1 1962 5 5 February-62 Ballot Tabulation Killie Notes 1 1 1962 6 6 February-62 A Message from the Board of Trustees Albert J. Klee Killie Notes 1 1 1962 7 7 February-62 Why Not Panchax Albert J. Klee Killie Notes 1 1 1962 8 10 February-62 Remarks on the Identification of Three Aphyosemions Albert J. Klee Killie Notes 1 1 1962 11 11 February-62 Flash... Just in from New York City Killie Notes 1 1 1962 12 12 February-62 Help for Beginning Killie fanciers Killie Notes 1 1 1962 12 12 February-62 A few remarks on sending eggs Killie Notes 1 1 1962 12 12 February-62 Egg listings start in March Killie Notes 1 1 1962 13 13 February-62 Let's support the AKA Killie Notes 1 1 1962 13 13 February-62 Our new Roster Killie Notes 1 1 1962 13 14 February-62 Editorially speaking Killie Notes 1 1 1962 14 15 February-62 George Maier addresses Chicago Group Killie Notes 1 1 1962 15 15 February-62 Wamted for research Purposes -Cubanichthys cubanensis Neal R. Foster Killie Notes 1 2 1962 3 4 March-62 Report from your Board of Trustees Albert J. Klee Killie Notes 1 2 1962 5 7 March-62 The Egg Bank (N. -

Coping with Life on Land: Physiological, Biochemical, and Structural Mechanisms to Enhance Function in Amphibious Fishes
Coping with Life on Land: Physiological, Biochemical, and Structural Mechanisms to Enhance Function in Amphibious Fishes by Andy Joseph Turko A Thesis presented to The University of Guelph In partial fulfilment of requirements for the degree of Doctor of Philosophy in Integrative Biology Guelph, Ontario, Canada © Andy Joseph Turko, October 2018 ABSTRACT COPING WITH LIFE ON LAND: PHYSIOLOGICAL, BIOCHEMICAL, AND STRUCTURAL MECHANISMS TO ENHANCE FUNCTION IN AMPHIBIOUS FISHES Andy Joseph Turko Advisor: University of Guelph, 2018 Dr. Patricia A. Wright The invasion of land by fishes was one of the most dramatic transitions in the evolutionary history of vertebrates. In this thesis, I investigated how amphibious fishes cope with increased effective gravity and the inability to feed while out of water. In response to increased body weight on land (7 d), the gill skeleton of Kryptolebias marmoratus became stiffer, and I found increased abundance of many proteins typically associated with bone and cartilage growth in mammals. Conversely, there was no change in gill stiffness in the primitive ray-finned fish Polypterus senegalus after one week out of water, but after eight months the arches were significantly shorter and smaller. A similar pattern of gill reduction occurred during the tetrapod invasion of land, and my results suggest that genetic assimilation of gill plasticity could be an underlying mechanism. I also found proliferation of a gill inter-lamellar cell mass in P. senegalus out of water (7 d) that resembled gill remodelling in several other fishes, suggesting this may be an ancestral actinopterygian trait. Next, I tested the function of a calcified sheath that I discovered surrounding the gill filaments of >100 species of killifishes and some other percomorphs. -

Gene Expression After Freshwater Transfer in Gills and Opercular Epithelia of Killifish: Insight Into Divergent Mechanisms of Ion Transport Graham R
The Journal of Experimental Biology 208, 2719-2729 2719 Published by The Company of Biologists 2005 doi:10.1242/jeb.01688 Gene expression after freshwater transfer in gills and opercular epithelia of killifish: insight into divergent mechanisms of ion transport Graham R. Scott1,*, James B. Claiborne2, Susan L. Edwards2,3, Patricia M. Schulte1 and Chris M. Wood4 1Department of Zoology, University of British Columbia, Vancouver BC, Canada V6T 1Z4, 2Department of Biology, Georgia Southern University, Statesboro, GA 30460-8042, USA, 3Department of Physiology and Pharmacology, James Cook University, Cairns, QLD 4879, Australia and 4Department of Biology, McMaster University, Hamilton ON, Canada L8S 4K1 *Author for correspondence (e-mail: [email protected]) Accepted 17 May 2005 Summary We have explored the molecular basis for differences in epithelium, NHE2 was not expressed; furthermore, physiological function between the gills and opercular Na+,K+-ATPase activity was unchanged after transfer to epithelium of the euryhaline killifish Fundulus freshwater, CA2 mRNA levels decreased, and NHE3 levels heteroclitus. These tissues are functionally similar in increased. Consistent with their functional similarities in seawater, but in freshwater the gills actively absorb Na+ seawater, killifish gills and opercular epithelium expressed – + + + + – but not Cl , whereas the opercular epithelium actively Na ,K -ATPase α1a, Na ,K ,2Cl cotransporter 1 absorbs Cl– but not Na+. These differences in freshwater (NKCC1), cystic fibrosis transmembrane conductance physiology are likely due to differences in absolute levels regulator (CFTR) Cl– channel and the signalling protein of gene expression (measured using real-time PCR), as 14-3-3a at similar absolute levels. Furthermore, NKCC1 several proteins important for Na+ transport, namely and CFTR were suppressed equally in each tissue after Na+,H+-exchanger 2 (NHE2), carbonic anhydrase 2 (CA2), freshwater transfer, and 14-3-3a mRNA increased in both. -
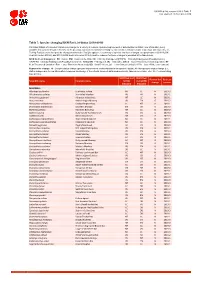
Table 7: Species Changing IUCN Red List Status (2018-2019)
IUCN Red List version 2019-3: Table 7 Last Updated: 10 December 2019 Table 7: Species changing IUCN Red List Status (2018-2019) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2018 (IUCN Red List version 2018-2) and 2019 (IUCN Red List version 2019-3) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered [CR(PE) - Critically Endangered (Possibly Extinct), CR(PEW) - Critically Endangered (Possibly Extinct in the Wild)], EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2018) List (2019) change version Category