Bioinformatics Interpretation of Exome Sequencing: Blood Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

High-Density P300 Enhancers Control Cell State Transitions Steven Witte1,2, Allan Bradley2, Anton J
Witte et al. BMC Genomics (2015) 16:903 DOI 10.1186/s12864-015-1905-6 RESEARCH ARTICLE Open Access High-density P300 enhancers control cell state transitions Steven Witte1,2, Allan Bradley2, Anton J. Enright3* and Stefan A. Muljo1* Abstract Background: Transcriptional enhancers are frequently bound by a set of transcription factors that collaborate to activate lineage-specific gene expression. Recently, it was appreciated that a subset of enhancers comprise extended clusters dubbed stretch- or super-enhancers (SEs). These SEs are located near key cell identity genes, and enriched for non-coding genetic variations associated with disease. Previously, SEs have been defined as having the highest density of Med1, Brd4 or H3K27ac by ChIP-seq. The histone acetyltransferase P300 has been used as a marker of enhancers, but little is known about its binding to SEs. Results: We establish that P300 marks a similar SE repertoire in embryonic stem cells as previously reported using Med1 and H3K27ac. We also exemplify a role for SEs in mouse T helper cell fate decision. Similarly, upon activation of macrophages by bacterial endotoxin, we found that many SE-associated genes encode inflammatory proteins that are strongly up-regulated. These SEs arise from small, low-density enhancers in unstimulated macrophages. We also identified expression quantitative trait loci (eQTL) in human monocytes that lie within such SEs. In macrophages and Th17 cells, inflammatory SEs can be perturbed either genetically or pharmacologically thus revealing new avenues to target inflammation. Conclusions: Our findings support the notion that P300-marked SEs can help identify key nodes of transcriptional control during cell fate decisions. -
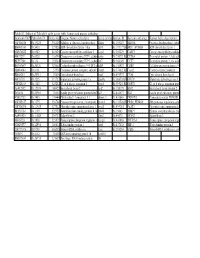
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

A Rare Variant in MCF2L Identified Using Exclusion Linkage in A
European Journal of Human Genetics (2016) 24, 86–91 & 2016 Macmillan Publishers Limited All rights reserved 1018-4813/16 www.nature.com/ejhg ARTICLE A rare variant in MCF2L identified using exclusion linkage in a pedigree with premature atherosclerosis Stephanie Maiwald1,7, Mahdi M Motazacker1,7,8, Julian C van Capelleveen1, Suthesh Sivapalaratnam1, Allard C van der Wal2, Chris van der Loos2, John JP Kastelein1, Willem H Ouwehand3,4, G Kees Hovingh1, Mieke D Trip1,5, Jaap D van Buul6 and Geesje M Dallinga-Thie*,1 Cardiovascular disease (CVD) is a major cause of death in Western societies. CVD risk is largely genetically determined. The molecular pathology is, however, not elucidated in a large number of families suffering from CVD. We applied exclusion linkage analysis and next-generation sequencing to elucidate the molecular defect underlying premature CVD in a small pedigree, comprising two generations of which six members suffered from premature CVD. A total of three variants showed co-segregation with the disease status in the family. Two of these variants were excluded from further analysis based on the prevalence in replication cohorts, whereas a non-synonymous variant in MCF.2 Cell Line Derived Transforming Sequence-like protein (MCF2L, c.2066A4G; p.(Asp689Gly); NM_001112732.1), located in the DH domain, was only present in the studied family. MCF2L is a guanine exchange factor that potentially links pathways that signal through Rac1 and RhoA. Indeed, in HeLa cells, MCF2L689Gly failed to activate Rac1 as well as RhoA, resulting in impaired stress fiber formation. Moreover, MCF2L protein was found in human atherosclerotic lesions but not in healthy tissue segments. -

Expression of the POTE Gene Family in Human Ovarian Cancer Carter J Barger1,2, Wa Zhang1,2, Ashok Sharma 1,2, Linda Chee1,2, Smitha R
www.nature.com/scientificreports OPEN Expression of the POTE gene family in human ovarian cancer Carter J Barger1,2, Wa Zhang1,2, Ashok Sharma 1,2, Linda Chee1,2, Smitha R. James3, Christina N. Kufel3, Austin Miller 4, Jane Meza5, Ronny Drapkin 6, Kunle Odunsi7,8,9, 2,10 1,2,3 Received: 5 July 2018 David Klinkebiel & Adam R. Karpf Accepted: 7 November 2018 The POTE family includes 14 genes in three phylogenetic groups. We determined POTE mRNA Published: xx xx xxxx expression in normal tissues, epithelial ovarian and high-grade serous ovarian cancer (EOC, HGSC), and pan-cancer, and determined the relationship of POTE expression to ovarian cancer clinicopathology. Groups 1 & 2 POTEs showed testis-specifc expression in normal tissues, consistent with assignment as cancer-testis antigens (CTAs), while Group 3 POTEs were expressed in several normal tissues, indicating they are not CTAs. Pan-POTE and individual POTEs showed signifcantly elevated expression in EOC and HGSC compared to normal controls. Pan-POTE correlated with increased stage, grade, and the HGSC subtype. Select individual POTEs showed increased expression in recurrent HGSC, and POTEE specifcally associated with reduced HGSC OS. Consistent with tumors, EOC cell lines had signifcantly elevated Pan-POTE compared to OSE and FTE cells. Notably, Group 1 & 2 POTEs (POTEs A/B/B2/C/D), Group 3 POTE-actin genes (POTEs E/F/I/J/KP), and other Group 3 POTEs (POTEs G/H/M) show within-group correlated expression, and pan-cancer analyses of tumors and cell lines confrmed this relationship. Based on their restricted expression in normal tissues and increased expression and association with poor prognosis in ovarian cancer, POTEs are potential oncogenes and therapeutic targets in this malignancy. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Genome Sequencing Unveils a Regulatory Landscape of Platelet Reactivity
ARTICLE https://doi.org/10.1038/s41467-021-23470-9 OPEN Genome sequencing unveils a regulatory landscape of platelet reactivity Ali R. Keramati 1,2,124, Ming-Huei Chen3,4,124, Benjamin A. T. Rodriguez3,4,5,124, Lisa R. Yanek 2,6, Arunoday Bhan7, Brady J. Gaynor 8,9, Kathleen Ryan8,9, Jennifer A. Brody 10, Xue Zhong11, Qiang Wei12, NHLBI Trans-Omics for Precision (TOPMed) Consortium*, Kai Kammers13, Kanika Kanchan14, Kruthika Iyer14, Madeline H. Kowalski15, Achilleas N. Pitsillides4,16, L. Adrienne Cupples 4,16, Bingshan Li 12, Thorsten M. Schlaeger7, Alan R. Shuldiner9, Jeffrey R. O’Connell8,9, Ingo Ruczinski17, Braxton D. Mitchell 8,9, ✉ Nauder Faraday2,18, Margaret A. Taub17, Lewis C. Becker1,2, Joshua P. Lewis 8,9,125 , 2,14,125✉ 3,4,125✉ 1234567890():,; Rasika A. Mathias & Andrew D. Johnson Platelet aggregation at the site of atherosclerotic vascular injury is the underlying patho- physiology of myocardial infarction and stroke. To build upon prior GWAS, here we report on 16 loci identified through a whole genome sequencing (WGS) approach in 3,855 NHLBI Trans-Omics for Precision Medicine (TOPMed) participants deeply phenotyped for platelet aggregation. We identify the RGS18 locus, which encodes a myeloerythroid lineage-specific regulator of G-protein signaling that co-localizes with expression quantitative trait loci (eQTL) signatures for RGS18 expression in platelets. Gene-based approaches implicate the SVEP1 gene, a known contributor of coronary artery disease risk. Sentinel variants at RGS18 and PEAR1 are associated with thrombosis risk and increased gastrointestinal bleeding risk, respectively. Our WGS findings add to previously identified GWAS loci, provide insights regarding the mechanism(s) by which genetics may influence cardiovascular disease risk, and underscore the importance of rare variant and regulatory approaches to identifying loci contributing to complex phenotypes. -

Different Species Common Arthritis
High Resolution Mapping of Cia3: A Common Arthritis Quantitative Trait Loci in Different Species This information is current as Xinhua Yu, Haidong Teng, Andreia Marques, Farahnaz of September 27, 2021. Ashgari and Saleh M. Ibrahim J Immunol 2009; 182:3016-3023; ; doi: 10.4049/jimmunol.0803005 http://www.jimmunol.org/content/182/5/3016 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2009/02/18/182.5.3016.DC1 Material References This article cites 36 articles, 9 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/182/5/3016.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology High Resolution Mapping of Cia3: A Common Arthritis Quantitative Trait Loci in Different Species1 Xinhua Yu, Haidong Teng, Andreia Marques, Farahnaz Ashgari, and Saleh M. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

New Breast Cancer Risk Variant Discovered at 10Q25 in East Asian Women
Published OnlineFirst May 15, 2013; DOI: 10.1158/1055-9965.EPI-12-1393 Cancer Epidemiology, Research Article Biomarkers & Prevention New Breast Cancer Risk Variant Discovered at 10q25 in East Asian Women Jiajun Shi1, Hyuna Sung2, Ben Zhang1, Wei Lu9, Ji-Yeob Choi2,5, Yong-Bing Xiang10, Mi Kyung Kim8,11, Motoki Iwasaki12, Jirong Long1, Bu-Tian Ji13, Sue K. Park2,3,5, Ying Zheng9, Shoichiro Tsugane12, Keun-Young Yoo3, Wenjing Wang9, Dong-Young Noh4,5, Wonshik Han4,5, Sung-Won Kim6, Min Hyuk Lee7, Jong Won Lee8, Jong-Young Lee14, Chen-Yang Shen15,16, Keitaro Matsuo17, Sei-Hyun Ahn8,11, Yu-Tang Gao10, Xiao Ou Shu1, Qiuyin Cai1, Daehee Kang2,3,5, and Wei Zheng1 Abstract Background: Recently, 41 new genetic susceptibility loci for breast cancer risk were identified in a genome- wide association study (GWAS) conducted in European descendants. Most of these risk variants have not been directly replicated in Asian populations. Methods: We evaluated nine of those nonreplication loci in East Asians to identify new risk variants for breast cancer in these regions. First, we analyzed single-nucleotide polymorphisms (SNP) in these regions using data from two GWAS conducted among Chinese and Korean women, including 5,083 cases and 4,376 controls (stage 1). In each region, we selected an SNP showing the strongest association with breast cancer risk for replication in an independent set of 7,294 cases and 9,404 controls of East Asian descents (stage 2). Logistic regression models were used to calculate adjusted ORs and 95% confidence intervals (CI) as a measure of the association of breast cancer risk and genetic variants. -

Thrombocytopenia-Associated Mutations in Ser/Thr Kinase MASTL Deregulate Actin Cytoskeletal Dynamics in Platelets
Thrombocytopenia-associated mutations in Ser/Thr kinase MASTL deregulate actin cytoskeletal dynamics in platelets Begoña Hurtado, … , Pablo García de Frutos, Marcos Malumbres J Clin Invest. 2018;128(12):5351-5367. https://doi.org/10.1172/JCI121876. Research Article Cell biology Hematology Graphical abstract Find the latest version: https://jci.me/121876/pdf The Journal of Clinical Investigation RESEARCH ARTICLE Thrombocytopenia-associated mutations in Ser/Thr kinase MASTL deregulate actin cytoskeletal dynamics in platelets Begoña Hurtado,1,2 Marianna Trakala,1 Pilar Ximénez-Embún,3 Aicha El Bakkali,1 David Partida,1 Belén Sanz-Castillo,1 Mónica Álvarez-Fernández,1 María Maroto,1 Ruth Sánchez-Martínez,1 Lola Martínez4, Javier Muñoz,3 Pablo García de Frutos,2 and Marcos Malumbres1 1Cell Division and Cancer Group, Spanish National Cancer Research Centre (CNIO), Madrid, Spain. 2Department of Cell Death and Proliferation, Institut d’Investigacions Biomèdiques de Barcelona-Consejo Superior de Investigaciones Científicas- Institut d’Investigacions Biomèdiques August Pi i Sunyer- (IIBB-CSIC-IDIBAPS), Barcelona, Spain. 3ProteoRed – Instituto de Salud Carlos III (ISCIII) and Proteomics Unit, CNIO, Madrid, Spain. 4Cytometry Unit, CNIO, Madrid, Spain. MASTL, a Ser/Thr kinase that inhibits PP2A-B55 complexes during mitosis, is mutated in autosomal dominant thrombocytopenia. However, the connections between the cell-cycle machinery and this human disease remain unexplored. We report here that, whereas Mastl ablation in megakaryocytes prevented proper maturation of these cells, mice carrying the thrombocytopenia-associated mutation developed thrombocytopenia as a consequence of aberrant activation and survival of platelets. Activation of mutant platelets was characterized by hyperstabilized pseudopods mimicking the effect of PP2A inhibition and actin polymerization defects.