A Temporally Controlled Sequence of X-Chromosome Inactivation and Reactivation Defines Female Mouse in Vitro Germ Cells with Meiotic Potential
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exceptional Conservation of Horse–Human Gene Order on X Chromosome Revealed by High-Resolution Radiation Hybrid Mapping
Exceptional conservation of horse–human gene order on X chromosome revealed by high-resolution radiation hybrid mapping Terje Raudsepp*†, Eun-Joon Lee*†, Srinivas R. Kata‡, Candice Brinkmeyer*, James R. Mickelson§, Loren C. Skow*, James E. Womack‡, and Bhanu P. Chowdhary*¶ʈ *Department of Veterinary Anatomy and Public Health, ‡Department of Veterinary Pathobiology, College of Veterinary Medicine, and ¶Department of Animal Science, College of Agriculture and Life Science, Texas A&M University, College Station, TX 77843; and §Department of Veterinary Pathobiology, University of Minnesota, 295f AS͞VM, St. Paul, MN 55108 Contributed by James E. Womack, December 30, 2003 Development of a dense map of the horse genome is key to efforts ciated with the traits, once they are mapped by genetic linkage aimed at identifying genes controlling health, reproduction, and analyses with highly polymorphic markers. performance. We herein report a high-resolution gene map of the The X chromosome is the most conserved mammalian chro- horse (Equus caballus) X chromosome (ECAX) generated by devel- mosome (18, 19). Extensive comparisons of structure, organi- oping and typing 116 gene-specific and 12 short tandem repeat zation, and gene content of this chromosome in evolutionarily -markers on the 5,000-rad horse ؋ hamster whole-genome radia- diverse mammals have revealed a remarkable degree of conser tion hybrid panel and mapping 29 gene loci by fluorescence in situ vation (20–22). Until now, the chromosome has been best hybridization. The human X chromosome sequence was used as a studied in humans and mice, where the focus of research has template to select genes at 1-Mb intervals to develop equine been the intriguing patterns of X inactivation and the involve- orthologs. -

Linked Mental Retardation Detected by Array CGH
JMG Online First, published on September 16, 2005 as 10.1136/jmg.2005.036178 J Med Genet: first published as 10.1136/jmg.2005.036178 on 16 September 2005. Downloaded from Chromosomal copy number changes in patients with non-syndromic X- linked mental retardation detected by array CGH D Lugtenberg1, A P M de Brouwer1, T Kleefstra1, A R Oudakker1, S G M Frints2, C T R M Schrander- Stumpel2, J P Fryns3, L R Jensen4, J Chelly5, C Moraine6, G Turner7, J A Veltman1, B C J Hamel1, B B A de Vries1, H van Bokhoven1, H G Yntema1 1Department of Human Genetics, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands; 2Department of Clinical Genetics, University Hospital Maastricht, Maastricht, The Netherlands; 3Center for Human Genetics, University of Leuven, Leuven, Belgium; 4Max Planck Institute for Molecular Genetics, Berlin, Germany; 5INSERM 129-ICGM, Faculté de Médecine Cochin, Paris, France; 6Service de Génétique et INSERM U316, Hôpital Bretonneau, Tours, France; 7 GOLD Program, Hunter Genetics, University of Newcastle, Callaghan, New South Wales 2308, Australia http://jmg.bmj.com/ Corresponding author: on October 2, 2021 by guest. Protected copyright. Helger G. Yntema, PhD Department of Human Genetics Radboud University Nijmegen Medical Centre P.O. Box 9101 6500 HB Nijmegen The Netherlands E-mail: [email protected] tel: +31-24-3613799 fax: +31-24-3616658 1 Copyright Article author (or their employer) 2005. Produced by BMJ Publishing Group Ltd under licence. J Med Genet: first published as 10.1136/jmg.2005.036178 on 16 September 2005. Downloaded from ABSTRACT Introduction: Several studies have shown that array based comparative genomic hybridization (array CGH) is a powerful tool for the detection of copy number changes in the genome of individuals with a congenital disorder. -

Nuclear Organization and the Epigenetic Landscape of the Mus Musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected]
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 8-9-2019 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia Liu University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Liu, Alicia, "Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome" (2019). Doctoral Dissertations. 2273. https://opencommons.uconn.edu/dissertations/2273 Nuclear Organization and the Epigenetic Landscape of the Mus musculus X-Chromosome Alicia J. Liu, Ph.D. University of Connecticut, 2019 ABSTRACT X-linked imprinted genes have been hypothesized to contribute parent-of-origin influences on social cognition. A cluster of imprinted genes Xlr3b, Xlr4b, and Xlr4c, implicated in cognitive defects, are maternally expressed and paternally silent in the murine brain. These genes defy classic mechanisms of autosomal imprinting, suggesting a novel method of imprinted gene regulation. Using Xlr3b and Xlr4c as bait, this study uses 4C-Seq on neonatal whole brain of a 39,XO mouse model, to provide the first in-depth analysis of chromatin dynamics surrounding an imprinted locus on the X-chromosome. Significant differences in long-range contacts exist be- tween XM and XP monosomic samples. In addition, XM interaction profiles contact a greater number of genes linked to cognitive impairment, abnormality of the nervous system, and abnormality of higher mental function. This is not a pattern that is unique to the imprinted Xlr3/4 locus. Additional Alicia J. Liu - University of Connecticut - 2019 4C-Seq experiments show that other genes on the X-chromosome, implicated in intellectual disability and/or ASD, also produce more maternal contacts to other X-linked genes linked to cognitive impairment. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Zbtb16 Regulates Social Cognitive Behaviors and Neocortical
Usui et al. Translational Psychiatry (2021) 11:242 https://doi.org/10.1038/s41398-021-01358-y Translational Psychiatry ARTICLE Open Access Zbtb16 regulates social cognitive behaviors and neocortical development Noriyoshi Usui 1,2,3,4, Stefano Berto5,AmiKonishi1, Makoto Kondo1,4, Genevieve Konopka5,HideoMatsuzaki 2,6,7 and Shoichi Shimada1,2,4 Abstract Zinc finger and BTB domain containing 16 (ZBTB16) play the roles in the neural progenitor cell proliferation and neuronal differentiation during development, however, how the function of ZBTB16 is involved in brain function and behaviors unknown. Here we show the deletion of Zbtb16 in mice leads to social impairment, repetitive behaviors, risk- taking behaviors, and cognitive impairment. To elucidate the mechanism underlying the behavioral phenotypes, we conducted histological analyses and observed impairments in thinning of neocortical layer 6 (L6) and a reduction of TBR1+ neurons in Zbtb16 KO mice. Furthermore, we found increased dendritic spines and microglia, as well as developmental defects in oligodendrocytes and neocortical myelination in the prefrontal cortex (PFC) of Zbtb16 KO mice. Using genomics approaches, we identified the Zbtb16 transcriptome that includes genes involved in neocortical maturation such as neurogenesis and myelination, and both autism spectrum disorder (ASD) and schizophrenia (SCZ) pathobiology. Co-expression networks further identified Zbtb16-correlated modules that are unique to ASD or SCZ, respectively. Our study provides insight into the novel roles of ZBTB16 in behaviors and neocortical development related to the disorders. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Introduction identified as a causative mutation for skeletal defects, ZBTB16 (PLZF) encodes a transcription factor, which genital hypoplasia, and mental retardation (SGYMR)6,7. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Centre for Arab Genomic Studies a Division of Sheikh Hamdan Award for Medical Sciences
Centre for Arab Genomic Studies A Division of Sheikh Hamdan Award for Medical Sciences The Catalogue for Transmission Genetics in Arabs CTGA Database tRNA Methyltransferase 1, S. Cerevisiae, Homolog of Alternative Names planus. These findings are further bolstered by the TRMT1 fact that mutations in two other RNA- N2,N2-Dimethylguanosine-26 tRNA methyltransferase genes, NSUN2 and FTSJ1, have Methyltransferase been associated with intellectual disability. tRNA(m(2,2)G26)Dimethyltransferase Record Category Molecular Genetics Gene locus The TRMT1 gene is located on the short arm of chromosome 19 and spans a length of 12.6 kb of WHO-ICD DNA. Its coding sequence is spread across 18 N/A to gene loci exons and it encodes a 72.2 kDa protein product comprised of 659 amino acids. An additional 69.3 Incidence per 100,000 Live Births kDa isoform of the TRMT1 protein exists due to an N/A to gene loci alternatively spliced transcript variant. The gene is widely expressed in the human body, particularly in OMIM Number the nervous system, intestine, spleen, kidney and 611669 lung. Mode of Inheritance Epidemiology in the Arab World N/A to gene loci Saudi Arabia Monies et al. (2017) studied the findings of 1000 Gene Map Locus diagnostic panels and exomes carried out at a next 19p13.13 generation sequencing lab in Saudi Arabia. One patient, a 13-year-old male, presented with speech Description delay, intellectual disability, learning disability, The TRMT1 gene encodes a methyltransferase hypotonia and seizures. Using whole exome enzyme that acts on tRNA. This enzyme, which sequencing, a homozygous mutation consists of a zinc finger motif and an (c.1245_1246del, p.L415fs) was identified in exon arginine/proline rich region at its C-terminus, is 10 of the patient’s TRMT1 gene. -
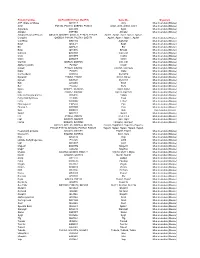
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

ITRAQ-Based Quantitative Proteomic Analysis of Processed Euphorbia Lathyris L
Zhang et al. Proteome Science (2018) 16:8 https://doi.org/10.1186/s12953-018-0136-6 RESEARCH Open Access ITRAQ-based quantitative proteomic analysis of processed Euphorbia lathyris L. for reducing the intestinal toxicity Yu Zhang1, Yingzi Wang1*, Shaojing Li2*, Xiuting Zhang1, Wenhua Li1, Shengxiu Luo1, Zhenyang Sun1 and Ruijie Nie1 Abstract Background: Euphorbia lathyris L., a Traditional Chinese medicine (TCM), is commonly used for the treatment of hydropsy, ascites, constipation, amenorrhea, and scabies. Semen Euphorbiae Pulveratum, which is another type of Euphorbia lathyris that is commonly used in TCM practice and is obtained by removing the oil from the seed that is called paozhi, has been known to ease diarrhea. Whereas, the mechanisms of reducing intestinal toxicity have not been clearly investigated yet. Methods: In this study, the isobaric tags for relative and absolute quantitation (iTRAQ) in combination with the liquid chromatography-tandem mass spectrometry (LC-MS/MS) proteomic method was applied to investigate the effects of Euphorbia lathyris L. on the protein expression involved in intestinal metabolism, in order to illustrate the potential attenuated mechanism of Euphorbia lathyris L. processing. Differentially expressed proteins (DEPs) in the intestine after treated with Semen Euphorbiae (SE), Semen Euphorbiae Pulveratum (SEP) and Euphorbiae Factor 1 (EFL1) were identified. The bioinformatics analysis including GO analysis, pathway analysis, and network analysis were done to analyze the key metabolic pathways underlying the attenuation mechanism through protein network in diarrhea. Western blot were performed to validate selected protein and the related pathways. Results: A number of differentially expressed proteins that may be associated with intestinal inflammation were identified. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

A Worldwide Map of Swine Short Tandem Repeats and Their
Wu et al. Genet Sel Evol (2021) 53:39 https://doi.org/10.1186/s12711-021-00631-4 Genetics Selection Evolution RESEARCH ARTICLE Open Access A worldwide map of swine short tandem repeats and their associations with evolutionary and environmental adaptations Zhongzi Wu1, Huanfa Gong1, Mingpeng Zhang1, Xinkai Tong1, Huashui Ai1, Shijun Xiao1, Miguel Perez‑Enciso2,3, Bin Yang1* and Lusheng Huang1* Abstract Background: Short tandem repeats (STRs) are genetic markers with a greater mutation rate than single nucleotide polymorphisms (SNPs) and are widely used in genetic studies and forensics. However, most studies in pigs have focused only on SNPs or on a limited number of STRs. Results: This study screened 394 deep‑sequenced genomes from 22 domesticated pig breeds/populations world‑ wide, wild boars from both Europe and Asia, and numerous outgroup Suidaes, and identifed a set of 878,967 poly‑ morphic STRs (pSTRs), which represents the largest repository of pSTRs in pigs to date. We found multiple lines of evidence that pSTRs in coding regions were afected by purifying selection. The enrichment of trinucleotide pSTRs in coding sequences (CDS), 5′UTR and H3K4me3 regions suggests that trinucleotide STRs serve as important com‑ ponents in the exons and promoters of the corresponding genes. We demonstrated that, compared to SNPs, pSTRs provide comparable or even greater accuracy in determining the breed identity of individuals. We identifed pSTRs that showed signifcant population diferentiation between domestic pigs and wild boars in Asia and Europe. We also observed that some pSTRs were signifcantly associated with environmental variables, such as average annual tem‑ perature or altitude of the originating sites of Chinese indigenous breeds, among which we identifed loss‑of‑function and/or expanded STRs overlapping with genes such as AHR, LAS1L and PDK1.