L1TD1 Sirna (H): Sc-88192
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

High-Density P300 Enhancers Control Cell State Transitions Steven Witte1,2, Allan Bradley2, Anton J
Witte et al. BMC Genomics (2015) 16:903 DOI 10.1186/s12864-015-1905-6 RESEARCH ARTICLE Open Access High-density P300 enhancers control cell state transitions Steven Witte1,2, Allan Bradley2, Anton J. Enright3* and Stefan A. Muljo1* Abstract Background: Transcriptional enhancers are frequently bound by a set of transcription factors that collaborate to activate lineage-specific gene expression. Recently, it was appreciated that a subset of enhancers comprise extended clusters dubbed stretch- or super-enhancers (SEs). These SEs are located near key cell identity genes, and enriched for non-coding genetic variations associated with disease. Previously, SEs have been defined as having the highest density of Med1, Brd4 or H3K27ac by ChIP-seq. The histone acetyltransferase P300 has been used as a marker of enhancers, but little is known about its binding to SEs. Results: We establish that P300 marks a similar SE repertoire in embryonic stem cells as previously reported using Med1 and H3K27ac. We also exemplify a role for SEs in mouse T helper cell fate decision. Similarly, upon activation of macrophages by bacterial endotoxin, we found that many SE-associated genes encode inflammatory proteins that are strongly up-regulated. These SEs arise from small, low-density enhancers in unstimulated macrophages. We also identified expression quantitative trait loci (eQTL) in human monocytes that lie within such SEs. In macrophages and Th17 cells, inflammatory SEs can be perturbed either genetically or pharmacologically thus revealing new avenues to target inflammation. Conclusions: Our findings support the notion that P300-marked SEs can help identify key nodes of transcriptional control during cell fate decisions. -

The Application of Mouse and Human Embryonic Stem Cells with Transcriptomics in Alternative Developmental Toxicity Tests
The application of mouse and human embryonic stem cells with transcriptomics in alternative developmental toxicity tests A bridge from model species to man Sjors Schulpen © Copyright All rights reserved. No part of this publication may be reproduced or transmitted in any form by any means without permission of the author. ISBN: 978-94-6203-861-5 Cover and layout: Maud van Deursen, Utrecht the Netherlands Production: CPI/ Wöhrmann Print Service, Zutphen the Netherlands The application of mouse and human embryonic stem cells with transcriptomics in alternative developmental toxicity tests A bridge from model species to man De toepassing van embryonale stamcellen van muis en mens met transcriptomics in alternatieve testen voor ontwikkelingstoxiciteit Een brug van modelorganisme naar de mens (met een samenvatting in het Nederlands) Proefschrift ter verkrijging van de graad van doctor aan de Universiteit Utrecht op gezag van de rector magnificus, prof.dr. G.J. van der Zwaan, ingevolge het besluit van het college voor promoties in het openbaar te verdedigen op dinsdag 7 juli 2015 des middags te 2.30 uur door Sjors Hubertus Wilhelmina Schulpen geboren op 5 januari 1983 te Sittard Promotoren: Prof. dr. A.H. Piersma Prof. dr. M. van den Berg Dit proefschrift werd mogelijk gemaakt door financiële steun van: Institute for Risk Assessment Sciences van de Universiteit Utrecht, Laboratorium voor gezondheidsbeschermingsonderzoek van het Rijksinstituut voor Volksgezondheid en Mileu, Greiner Bio-One en Stichting Proefdiervrij. Contents Abbreviations 6 -

Identification of Functional Genetic Variants in Inflammatory Bowel Disease by Genome and Transcriptome Sequencing
Identification of Functional Genetic Variants in Inflammatory Bowel Disease by Genome and Transcriptome Sequencing Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Christian-Albrechts-Universität zu Kiel vorgelegt von Matthias Barann August, 2012 Kiel, Deutschland 1 2 Referent/in: Prof. Dr. Philip Rosenstiel Korreferent/in: Prof. Dr. Dr. h.c. Thomas Bosch Tag der mündlichen Prüfung: 31.10.2012 Zum Druck genehmigt: 31.10.2012 gez. Prof. Dr. Wolfgang J. Duschl, Dekan 3 Parts of this dissertation are contained in the following two manuscripts. Rosenstiel R, Barann M, Klostermeier UC, Sheth V, Ellinghaus D, Rausch T, Korbel J, Nothnagel M, Krawczak M, Gilissen C, Veltman J, Forster M, Stade B, McLaughlin S, Lee CC, Fritscher-Ravens A, Franke A, Schreiber S. Whole Genome Sequence of a Crohn disease trio Barann M, Klostermeier UC, Esser D, Ammerpohl O, Siebert R, Sudbrak R, Lehrach H, Schreiber S, Rosenstiel R. Janus – Investigating the two faces of transcription 4 Contents 1. Introduction ............................................................................................................................. 7 1.1. Pathogenesis of inflammatory bowel diseases ...................................................................................... 8 1.2. Physical barrier function of the gut ......................................................................................................... 11 1.3. Pathogen recognition .................................................................................................................................. -

Repurposing Non-Invasive Prenatal Testing Data; Population Study of Single Nucleotide Variants Associated with Colorectal Cancer and Lynch Syndrome
Repurposing Non-invasive prenatal testing data; population study of single nucleotide variants associated with colorectal cancer and Lynch syndrome Natalia Forgacova ( [email protected] ) Comenius University in Bratislava Faculty of Natural Sciences: Univerzita Komenskeho v Bratislave Prirodovedecka fakulta https://orcid.org/0000-0001-6764-0895 Juraj Gazdarica Comenius University Science Park, Bratislava Jaroslav Budis Comenius University Science Park, Bratislava Jan Radvanszky Comenius University Science Park, Bratislava Tomas Szemes Comenius University Science Park, Bratislava Research Article Keywords: Colorectal cancer, Lynch syndrome, Non-invasive prenatal testing, Low-coverage massively parallel whole genome sequencing Posted Date: March 13th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-286309/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/26 Abstract PURPOSE: In our previous work, we described genomic data generated through non-invasive prenatal testing (NIPT) based on low-coverage massively parallel whole-genome sequencing of total plasma DNA of pregnant women in Slovakia as a valuable source of population specic data. In the present study, we used these data to determine the population allele frequency of common risk variants located in genes associated with colorectal cancer (CRC) and Lynch syndrome (LS). METHODS: Allele frequencies of identied variants were compared with six world populations to detect signicant differences between populations. Finally, we interpreted variants and searched for functional consequences and clinical signicance of variants using publicly available databases. RESULTS: Although we could not identify any pathogenic variants associated with CRC or LS in the Slovak population using NIPT data, we observed signicant differences in the allelic frequency of risk CRC variants previously reported in GWAS and common variants located in genes associated with LS. -
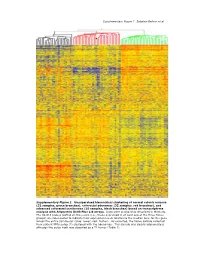
Supplementary Data
Supplementary Figure 1. Sabates-Bellver et al. * Supplementary Figure 1. Unsupervised hierarchical clustering of normal colonic mucosa (32 samples, green branches), colorectal adenomas (32 samples, red branches), and advanced colorectal carcinomas (25 samples, black branches) based on transcriptome analysis with Affymetrix U133 Plus 2.0 arrays. Data were analyzed as described in Methods. The 32,813 probes plotted on the y-axis (i.e., those expressed in at least one of the three tissue groups) are color-coded to indicate their expression levels relative to the median level for the gene across the entire sample set (blue: lower; red: higher). As expected, the tissue sample collected from patient NM’s polyp (*) clustered with the adenomas. This sample was clearly adenomatous although the polyp itself was classified as a T1 tumor (Table 1). Supplementary Figure 2; Sabates-Bellver et al. A B mitosis humoral defense mechanism CDC25B CCR2 CDC2 CCL14 CCNF C7 RAN CCL14 CDCA5 TNFRSF17 MAD2L1 CLU MAD2L1 CR2 CCNB1 C1S PRMT5 TNFRSF7 BUB1B SERPING1 PBK CD53 TPX2 KLRK1 BUB1 LY9 CDC2 CSF1R MAD2L2 KLRC4 CDC20 CLEC2B CDC6 C1QC UBE2C C1QA NUSAP1 C1QB AURKA HHEX BUB1 C1S CDC2 KLRC1 CCNA2 KLRD1 HCAP-G ITGB2 PTTG1 SPINK5 SMC2L1 HHEX PLK1 CD163 CCNA2 CCR2 KNTC2 MAG NEK2 CD163 KIF2C C3 BRRN1 XCL1 CCNB2 XCL2 NOLC1 ITGB2 KIF23 KLRB1 HCAP-G SLAMF1 KIF2C KLRD1 RCC1 LY9 AURKA CLEC2D CCNB1 CD1C CDC25C CD53 KIF11 CR1 ASPM CD74 CDC6 IGSF2 NEK2 CLEC2D KIFC1 CLU CDC25C CLU CENPE CCL18 NUSAP1 KLRF1 CDC25A CCL18 CCNF CR1 CDC25A CD40 CDC2 CCR7 adenomas normals CCR6 adenomas normals C D inflammatory response inflammatory response IL8 CCR2 ALOX5 NR3C1 IL8 NR3C1 MIF NR3C1 CXCL11 CXCL12 CXCL3 NR3C1 RIPK2 NR3C1 TLR4 AOC3 CXCL1 CD14 CXCL11 TLR7 CXCL2 STAB1 PF4 CXCL13 TLR4 CCL23 TLR4 HDAC9 CCL20 CCL21 TLR4 CCL5 CCL24 CCL5 S100A8 LY86 ALOX5 ITGAL S100A9 CCL23 REG3A CCL8 ANXA1 STAB1 SERPINA3 CCL5 IL1B CXCL12 IL1B CCL19 CCL26 BDKRB1 adenomas normals IL1R1 SIGLEC1 ADORA3 CCL2 PREX1 TLR3 Supplementary Figure 2. -

Number 5 May 2014
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST -CNRS Volume 18 - Number 5 May 2014 The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific Research (CNRS) on its electronic publishing platform I-Revues. Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS. Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST -CNRS Scope The Atlas of Genetics and Cytogenetics in Oncology and Haematology is a peer reviewed on-line journal in open access, devoted to genes, cytogenetics, and clinical entities in cancer, and cancer-prone diseases. It presents structured review articles (“cards”) on genes, leukaemias, solid tumours, cancer-prone diseases, and also more traditional review articles (“deep insights”) on the above subjects and on surrounding topics. It also present case reports in hematology and educational items in the various related topics for students in Medicine and in Sciences. Editorial correspondance Jean-Loup Huret Genetics, Department of Medical Information, University Hospital F-86021 Poitiers, France tel +33 5 49 44 45 46 or +33 5 49 45 47 67 [email protected] or [email protected] Staff Mohammad Ahmad, Mélanie Arsaban, Marie-Christine Jacquemot-Perbal, Vanessa Le Berre, Anne Malo, Carol Moreau, Catherine Morel-Pair, Laurent Rassinoux, Alain Zasadzinski. Philippe Dessen is the Database Director, and Alain Bernheim the Chairman of the on-line version (Gustave Roussy Institute – Villejuif – France). -

Replication of Linkage at Chromosome 20P13 and Identification Of
Werling et al. Molecular Autism 2014, 5:13 http://www.molecularautism.com/content/5/1/13 RESEARCH Open Access Replication of linkage at chromosome 20p13 and identification of suggestive sex-differential risk loci for autism spectrum disorder Donna M Werling1, Jennifer K Lowe2,3,4, Rui Luo2,5, Rita M Cantor4,5 and Daniel H Geschwind2,3,4,5* Abstract Background: Autism spectrum disorders (ASDs) are male-biased and genetically heterogeneous. While sequencing of sporadic cases has identified de novo risk variants, the heritable genetic contribution and mechanisms driving the male bias are less understood. Here, we aimed to identify familial and sex-differential risk loci in the largest available, uniformly ascertained, densely genotyped sample of multiplex ASD families from the Autism Genetics Resource Exchange (AGRE), and to compare results with earlier findings from AGRE. Methods: From a total sample of 1,008 multiplex families, we performed genome-wide, non-parametric linkage analysis in a discovery sample of 847 families, and separately on subsets of families with only male, affected children (male-only, MO) or with at least one female, affected child (female-containing, FC). Loci showing evidence for suggestive linkage (logarithm of odds ≥2.2) in this discovery sample, or in previous AGRE samples, were re-evaluated in an extension study utilizing all 1,008 available families. For regions with genome-wide significant linkage signal in the discovery stage, those families not included in the corresponding discovery sample were then evaluated for independent replication of linkage. Association testing of common single nucleotide polymorphisms (SNPs) was also performed within suggestive linkage regions. -

Bioinformatics Interpretation of Exome Sequencing: Blood Cancer
pISSN 1598-866X eISSN 2234-0742 Genomics Inform 2013;11(1):24-33 G&I Genomics & Informatics http://dx.doi.org/10.5808/GI.2013.11.1.24 ORIGINAL ARTICLE Bioinformatics Interpretation of Exome Sequencing: Blood Cancer Jiwoong Kim1†, Yun-Gyeong Lee1,2†, Namshin Kim1,2* 1Korean Bioinformation Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea, 2Department of Bioinformatics, University of Science and Technology, Daejeon 305-806, Korea We had analyzed 10 exome sequencing data and single nucleotide polymorphism chips for blood cancer provided by the PGM21 (The National Project for Personalized Genomic Medicine) Award program. We had removed sample G06 because the pair is not correct and G10 because of possible contamination. In-house software somatic copy-number and heterozygosity alteration estimation (SCHALE) was used to detect one loss of heterozygosity region in G05. We had discovered 27 functionally important mutations. Network and pathway analyses gave us clues that NPM1, GATA2, and CEBPA were major driver genes. By comparing with previous somatic mutation profiles, we had concluded that the provided data originated from acute myeloid leukemia. Protein structure modeling showed that somatic mutations in IDH2, RASGEF1B, and MSH4 can affect protein structures. Keywords: acute myeloid leukemia, computational biology, exome, mutation Introduction We had used an in-house bioinformatics pipeline and databases for this research, especially “somatic copy-number Thanks to the next-generation sequencing (NGS) tech- and heterozygosity alteration estimation” (SCHALE) to find nology, genomics research area has been developed drama- LOH and SCNA. Furthermore, driver genes were also tically in recent years. Since 2008, many efforts have been inferred from network and pathway analyses. -

RNA-Seq Analysis Reveals Pluripotency-Associated Genes and Their Interaction Networks in Human Embryonic Stem Cells
bioRxiv preprint doi: https://doi.org/10.1101/782169; this version posted September 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. RNA-Seq analysis reveals pluripotency-associated genes and their interaction networks in human embryonic stem cells Arindam Ghosh and Anup Som* Centre of Bioinformatics, Institute of Interdisciplinary Studies, University of Allahabad. *Corresponding Author: Anup Som. Centre of Bioinformatics, Institute of Interdisciplinary Studies, University of Allahabad, Prayagraj – 211002, INDIA. E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/782169; this version posted September 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ABSTRACT Insight into the key genes of pluripotency in human and their interrelationships is necessary for understanding the underlying mechanism of pluripotency and hence their successful application in regenerative medicine. The recent advances in transcriptomics technologies have created new opportunities to decipher the genes involved in pluripotency, genetic network that governs the unique properties of embryonic stem cells and lineage differentiation mechanisms in a deeper scale. There are a large number of experimental studies on human embryonic stem cells (hESCs) being routinely conducted for unfolding the underlying biology of embryogenesis and their clinical prospects. -

S41467-021-25107-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-021-25107-3 OPEN OCT4 cooperates with distinct ATP-dependent chromatin remodelers in naïve and primed pluripotent states in human Xin Huang 1,14, Kyoung-mi Park2,3,14, Paul Gontarz2,3, Bo Zhang 2,3, Joshua Pan4,5, Zachary McKenzie4, Laura A. Fischer2,3, Chen Dong 2,3, Sabine Dietmann2,6, Xiaoyun Xing 3,7, Pavel V. Shliaha8, Jihong Yang 1, Dan Li 1,9, Junjun Ding 9,10, Tenzin Lungjangwa11, Maya Mitalipova11, Shafqat A. Khan2,3, Sumeth Imsoonthornruksa11,12, Nick Jensen 2,3, Ting Wang 3,7, Cigall Kadoch 4,5, Rudolf Jaenisch 11,13, ✉ ✉ Jianlong Wang 1 & Thorold W. Theunissen 2,3 1234567890():,; Understanding the molecular underpinnings of pluripotency is a prerequisite for optimal maintenance and application of embryonic stem cells (ESCs). While the protein-protein interactions of core pluripotency factors have been identified in mouse ESCs, their inter- actome in human ESCs (hESCs) has not to date been explored. Here we mapped the OCT4 interactomes in naïve and primed hESCs, revealing extensive connections to mammalian ATP-dependent nucleosome remodeling complexes. In naïve hESCs, OCT4 is associated with both BRG1 and BRM, the two paralog ATPases of the BAF complex. Genome-wide location analyses and genetic studies reveal that these two enzymes cooperate in a functionally redundant manner in the transcriptional regulation of blastocyst-specific genes. In contrast, in primed hESCs, OCT4 cooperates with BRG1 and SOX2 to promote chromatin accessibility at ectodermal genes. This work reveals how a common transcription factor utilizes differential BAF complexes to control distinct transcriptional programs in naïve and primed hESCs.