Exploiting Knowledge on Structure–Activity Relationships for Designing Peptidomimetics of Endogenous Peptides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

B Number Gene Name Mrna Intensity Mrna
sample) total list predicted B number Gene name assignment mRNA present mRNA intensity Gene description Protein detected - Membrane protein membrane sample detected (total list) Proteins detected - Functional category # of tryptic peptides # of tryptic peptides # of tryptic peptides detected (membrane b0002 thrA 13624 P 39 P 18 P(m) 2 aspartokinase I, homoserine dehydrogenase I Metabolism of small molecules b0003 thrB 6781 P 9 P 3 0 homoserine kinase Metabolism of small molecules b0004 thrC 15039 P 18 P 10 0 threonine synthase Metabolism of small molecules b0008 talB 20561 P 20 P 13 0 transaldolase B Metabolism of small molecules chaperone Hsp70; DNA biosynthesis; autoregulated heat shock b0014 dnaK 13283 P 32 P 23 0 proteins Cell processes b0015 dnaJ 4492 P 13 P 4 P(m) 1 chaperone with DnaK; heat shock protein Cell processes b0029 lytB 1331 P 16 P 2 0 control of stringent response; involved in penicillin tolerance Global functions b0032 carA 9312 P 14 P 8 0 carbamoyl-phosphate synthetase, glutamine (small) subunit Metabolism of small molecules b0033 carB 7656 P 48 P 17 0 carbamoyl-phosphate synthase large subunit Metabolism of small molecules b0048 folA 1588 P 7 P 1 0 dihydrofolate reductase type I; trimethoprim resistance Metabolism of small molecules peptidyl-prolyl cis-trans isomerase (PPIase), involved in maturation of b0053 surA 3825 P 19 P 4 P(m) 1 GenProt outer membrane proteins (1st module) Cell processes b0054 imp 2737 P 42 P 5 P(m) 5 GenProt organic solvent tolerance Cell processes b0071 leuD 4770 P 10 P 9 0 isopropylmalate -

(12) Patent Application Publication (10) Pub. No.: US 2006/0110747 A1 Ramseier Et Al
US 200601 10747A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0110747 A1 Ramseier et al. (43) Pub. Date: May 25, 2006 (54) PROCESS FOR IMPROVED PROTEIN (60) Provisional application No. 60/591489, filed on Jul. EXPRESSION BY STRAIN ENGINEERING 26, 2004. (75) Inventors: Thomas M. Ramseier, Poway, CA Publication Classification (US); Hongfan Jin, San Diego, CA (51) Int. Cl. (US); Charles H. Squires, Poway, CA CI2O I/68 (2006.01) (US) GOIN 33/53 (2006.01) CI2N 15/74 (2006.01) Correspondence Address: (52) U.S. Cl. ................................ 435/6: 435/7.1; 435/471 KING & SPALDING LLP 118O PEACHTREE STREET (57) ABSTRACT ATLANTA, GA 30309 (US) This invention is a process for improving the production levels of recombinant proteins or peptides or improving the (73) Assignee: Dow Global Technologies Inc., Midland, level of active recombinant proteins or peptides expressed in MI (US) host cells. The invention is a process of comparing two genetic profiles of a cell that expresses a recombinant (21) Appl. No.: 11/189,375 protein and modifying the cell to change the expression of a gene product that is upregulated in response to the recom (22) Filed: Jul. 26, 2005 binant protein expression. The process can improve protein production or can improve protein quality, for example, by Related U.S. Application Data increasing solubility of a recombinant protein. Patent Application Publication May 25, 2006 Sheet 1 of 15 US 2006/0110747 A1 Figure 1 09 010909070£020\,0 10°0 Patent Application Publication May 25, 2006 Sheet 2 of 15 US 2006/0110747 A1 Figure 2 Ester sers Custer || || || || || HH-I-H 1 H4 s a cisiers TT closers | | | | | | Ya S T RXFO 1961. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases
Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases Alan J. Barrett Neil D. Rawlings J. Fred Woessner Editor biographies xxi Contributors xxiii Preface xxxi Introduction ' Abbreviations xxxvii ASPARTIC PEPTIDASES Introduction 1 Aspartic peptidases and their clans 3 2 Catalytic pathway of aspartic peptidases 12 Clan AA Family Al 3 Pepsin A 19 4 Pepsin B 28 5 Chymosin 29 6 Cathepsin E 33 7 Gastricsin 38 8 Cathepsin D 43 9 Napsin A 52 10 Renin 54 11 Mouse submandibular renin 62 12 Memapsin 1 64 13 Memapsin 2 66 14 Plasmepsins 70 15 Plasmepsin II 73 16 Tick heme-binding aspartic proteinase 76 17 Phytepsin 77 18 Nepenthesin 85 19 Saccharopepsin 87 20 Neurosporapepsin 90 21 Acrocylindropepsin 9 1 22 Aspergillopepsin I 92 23 Penicillopepsin 99 24 Endothiapepsin 104 25 Rhizopuspepsin 108 26 Mucorpepsin 11 1 27 Polyporopepsin 113 28 Candidapepsin 115 29 Candiparapsin 120 30 Canditropsin 123 31 Syncephapepsin 125 32 Barrierpepsin 126 33 Yapsin 1 128 34 Yapsin 2 132 35 Yapsin A 133 36 Pregnancy-associated glycoproteins 135 37 Pepsin F 137 38 Rhodotorulapepsin 139 39 Cladosporopepsin 140 40 Pycnoporopepsin 141 Family A2 and others 41 Human immunodeficiency virus 1 retropepsin 144 42 Human immunodeficiency virus 2 retropepsin 154 43 Simian immunodeficiency virus retropepsin 158 44 Equine infectious anemia virus retropepsin 160 45 Rous sarcoma virus retropepsin and avian myeloblastosis virus retropepsin 163 46 Human T-cell leukemia virus type I (HTLV-I) retropepsin 166 47 Bovine leukemia virus retropepsin 169 48 -

Table 4. V. Cholerae Flexgene ORF Collection
Table 4. V. cholerae FLEXGene ORF collection Reference Clone protein PlasmID clone GenBank Locus tag Symbol accession identifier FLEX clone name accession Product name VC0001 NP_062585 VcCD00019918 FLH200476.01F DQ772770 hypothetical protein VC0002 mioC NP_062586 VcCD00019938 FLH200506.01F DQ772771 mioC protein VC0003 thdF NP_062587 VcCD00019958 FLH200531.01F DQ772772 thiophene and furan oxidation protein ThdF VC0004 yidC NP_062588 VcCD00019970 FLH200545.01F DQ772773 inner membrane protein, 60 kDa VC0005 NP_062589 VcCD00061243 FLH236482.01F DQ899316 conserved hypothetical protein VC0006 rnpA NP_062590 VcCD00025697 FLH214799.01F DQ772774 ribonuclease P protein component VC0007 rpmH NP_062591 VcCD00061229 FLH236450.01F DQ899317 ribosomal protein L34 VC0008 NP_062592 VcCD00019917 FLH200475.01F DQ772775 amino acid ABC transporter, ATP-binding protein VC0009 NP_062593 VcCD00019966 FLH200540.01F DQ772776 amino acid ABC transproter, permease protein VC0010 NP_062594 VcCD00019152 FLH199275.01F DQ772777 amino acid ABC transporter, periplasmic amino acid-binding portion VC0011 NP_062595 VcCD00019151 FLH199274.01F DQ772778 hypothetical protein VC0012 dnaA NP_062596 VcCD00017363 FLH174286.01F DQ772779 chromosomal DNA replication initiator DnaA VC0013 dnaN NP_062597 VcCD00017316 FLH174063.01F DQ772780 DNA polymerase III, beta chain VC0014 recF NP_062598 VcCD00019182 FLH199319.01F DQ772781 recF protein VC0015 gyrB NP_062599 VcCD00025458 FLH174642.01F DQ772782 DNA gyrase, subunit B VC0016 NP_229675 VcCD00019198 FLH199346.01F DQ772783 hypothetical protein -

Supplemental Table 1A. Differential Gene Expression Profile of Adehcd40l and Adehnull Treated Cells Vs Untreated Cells
Supplemental Table 1a. Differential Gene Expression Profile of AdEHCD40L and AdEHNull treated cells vs Untreated Cells Fold change Regulation Fold change Regulation ([AdEHCD40L] vs ([AdEHCD40L] ([AdEHNull] vs ([AdEHNull] vs Probe Set ID [Untreated]) vs [Untreated]) [Untreated]) [Untreated]) Gene Symbol Gene Title RefSeq Transcript ID NM_001039468 /// NM_001039469 /// NM_004954 /// 203942_s_at 2.02 down 1.00 down MARK2 MAP/microtubule affinity-regulating kinase 2 NM_017490 217985_s_at 2.09 down 1.00 down BAZ1A fibroblastbromodomain growth adjacent factor receptorto zinc finger 2 (bacteria-expressed domain, 1A kinase, keratinocyte NM_013448 /// NM_182648 growth factor receptor, craniofacial dysostosis 1, Crouzon syndrome, Pfeiffer 203638_s_at 2.10 down 1.01 down FGFR2 syndrome, Jackson-Weiss syndrome) NM_000141 /// NM_022970 1570445_a_at 2.07 down 1.01 down LOC643201 hypothetical protein LOC643201 XM_001716444 /// XM_001717933 /// XM_932161 231763_at 3.05 down 1.02 down POLR3A polymerase (RNA) III (DNA directed) polypeptide A, 155kDa NM_007055 1555368_x_at 2.08 down 1.04 down ZNF479 zinc finger protein 479 NM_033273 /// XM_001714591 /// XM_001719979 241627_x_at 2.15 down 1.05 down FLJ10357 hypothetical protein FLJ10357 NM_018071 223208_at 2.17 down 1.06 down KCTD10 potassium channel tetramerisation domain containing 10 NM_031954 219923_at 2.09 down 1.07 down TRIM45 tripartite motif-containing 45 NM_025188 242772_x_at 2.03 down 1.07 down Transcribed locus 233019_at 2.19 down 1.08 down CNOT7 CCR4-NOT transcription complex, subunit 7 NM_013354 -

The Penicillin-Binding Proteins: Structure and Role in Peptidoglycan
1 The Penicillin-Binding Proteins: Structure and Role in Peptidoglycan 2 Biosynthesis 3 4 5 Eric Sauvage 1* , Mohammed Terrak 1, Juan A. Ayala 2, Paulette Charlier 1 6 7 8 1Centre d’Ingénierie des Protéines, Institut de Physique B5a et Institut de Chimie B6a, University of 9 Liège, B-4000 Sart Tilman, Belgium 10 2Centro de Biologia Molecular « Severo Ochoa » CSIC-UAM, Campus de Cantoblanco, 28049 11 Madrid, Spain 12 13 Keywords: penicillin binding, transpeptidase, transglycosylase, peptidoglycan synthesis, 3D 14 structure 15 Running head: Structure and role of PBPs 16 17 *Corresponding author : 18 Eric Sauvage 19 1Centre d’Ingénierie des Protéines, Institut de Physique B5a et Institut de Chimie B6a, University of 20 Liège, B-4000 Sart Tilman, Belgium. Phone: #32 43663620, Fax: #32 43663748 21 22 23 1 1 Abstract 2 3 Penicillin-binding proteins (PBPs) have been scrutinized for over 40 years. Recent structural 4 information on PBPs together with the ongoing long-term biochemical experimental investigations, 5 and results from more recent techniques such as protein localization by GFP-fusion 6 immunofluorescence or double-hybrid assay, have brought our understanding of the last stages of 7 the peptidoglycan biosynthesis to an outstanding level that allows a broad outlook on the properties 8 of these enzymes. Details are emerging regarding the interaction between the peptidoglycan- 9 synthesizing PBPs and the peptidoglycan, their mesh net-like product that surrounds and protects 10 bacteria. This review focuses on the detailed structure of PBPs and their implication in 11 peptidoglycan synthesis, maturation and recycling. An overview of the content in PBPs of some 12 bacteria is provided with an emphasis on comparing the biochemical properties of homologous 13 PBPs (orthologues) belonging to different bacteria. -

Structural Features of Angiotensin-I Converting Enzyme Catalytic Sites
Current Topics in Medicinal Chemistry 2004, 4, 403-429 403 Structural Features of Angiotensin-I Converting Enzyme Catalytic Sites: Conformational Studies in Solution, Homology Models and Comparison with Other Zinc Metallopeptidases Georgios A. Spyroulias*,1, Athanassios S. Galanis1, George Pairas1, Evy Manessi-Zoupa2 and Paul Cordopatis*,1 Departments of Pharmacy1 and Chemistry2, University of Patras, GR-26504, Patras, GREECE Abstract: Angiotensin-I Converting Enzyme (ACE) is a Zinc Metallopeptidase of which the three-dimensional stucture was unknown until recently, when the Xray structure of testis isoform (C-terminal domain of somatic) was determined. ACE plays an important role in the regulation of blood pressure due to its action in the frame of the Renin-Angiotensin System. Efforts for the specific inhibition of the catalytic function of this enzyme have been made on the basis of the X- ray structures of other enzymes with analogous efficacy in the hydrolytic cleavage of peptide substrate terminal fragments. Angiotensin-I Converting Enzyme bears the sequence and topology characteristics of the well-known gluzincins, a sub-family of zincins metallopeptidases and these similarities are exploited in order to reveal common structural elements among these enzymes. 3D homology models are also built using the X-ray structure of Thermolysin as template and peptide models that represent the amino acid sequence of the ACE’s two catalytic, zinc-containing sites are designed and synthesized. Conformational analysis of the zinc-free and zinc-bound peptides through high resolution 1H NMR Spectroscopy provides new insights into the solution structure of ACE catalytic centers. Structural properties of these peptides could provide valuable information towards the design and preparation of new potent ACE inhibitors. -

Polyamines Mitigate Antibiotic Inhibition of A.Actinomycetemcomitans Growth
Polyamines Mitigate Antibiotic Inhibition of A.actinomycetemcomitans Growth THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Allan Wattimena Graduate Program in Dentistry The Ohio State University 2017 Master's Examination Committee: Dr John Walters, Advisor Dr Purnima Kumar Dr Sara Palmer Dr Shareef Dabdoub Copyright by Allan Wattimena 2017 Abstract Polyamines are ubiquitous polycationic molecules that are present in all prokaryotic and eukaryotic cells. They are the breakdown products of amino acids and are important modulators of cell growth, stress and cell proliferation. Polyamines are present in higher concentrations in the periodontal pocket and may affect antibiotic resistance of bacterial biofilms. The effect of polyamines was investigated with amoxicillin (AMX), azithromycin (AZM) and doxycycline (DOX) on the growth of Aggregatibacter actinomycetemcomitans (A.a.) Y4 strain. Bacteria were grown in brain heart infusion broth under the following conditions: 1) A.a. only, 2) A.a. + antibiotic, 3) A.a. + antibiotic + polyamine mix (1.4mM putrescine, 0.4mM spermidine, 0.4mM spermine). Growth curve analysis, MIC determination and metatranscriptomic analysis were carried out. The presence of exogenous polyamines produced a small, but significant increase in growth of A.a. Polyamines mitigated the inhibitory effect of AMX, AZM and DOX on A.a. growth. Metatranscriptomic analysis revealed differing transcriptomic profiles when comparing AMX and AZM in the presence of polyamines. Polyamines produced a transient mitigation of AMX inhibition, but did not have a significant effect on gene transcription. Many gene transcription changes were seen when polyamines were in the presence of AZM. -

Inhibition of Bacterial Growthby ,3-Chloro-D-Alanine
Proc. Nat. Acad. Sci. USA Vol. 71, No. 2, pp. 417-421,.February 1974 Inhibition of Bacterial Growth by ,3-Chloro-D-Alanine (cell wall/alanine racemase/n-amino acids) JAMES M. MANNING, NANCY E. MERRIFIELD, WANDA M. JONES, AND EMIL C. GOTSCHLICH The Rockefeller University, New York, N.Y. 10021 Communicated by Alton Meister, October 3, 2973 ABSTRACT The D- and L-isomers of O-chloroalanine C, 22.50; H, 4.49; N, 8.75). At pH 7.4 and 370 in potassium inhibit the growth of Diplococcus pneumoniae, Strep- phosphate, (3-chloroalanine has a half-life of 30 hr. D-alanine, tococcus pyogenes, Bacillus subtilis, and Escherichia coli. With pneumococcus the inhibition by ,8-chloro-D-alanine pyridoxal 5'-phosphate, and crystalline suspensions of lactate is completely prevented by either D-alanine or D-alanyl- dehydrogenase (EC 1.1.1.27; ilactate:NAD+ oxidoreduc- D-alanine, while L-alanine is not effective in preventing the tase) and of glutamate dehydrogenase [EC 1.4.1.2; -gluta- inhibition. The inhibition of growth by (-chloro-L-alanine mate: NAD+ oxidoreductase(deaminating)] in saturated is not affected by D-alanine and is only partially prevented was by high concentrations of L-alanine. The intracellular (NH4)2SO4 were obtained from Sigma. D-Alanyl-D-alanine free alanine in untreated E. coli and B. subtilis is about a product of Fox Chemical Co. The strain of Bacillus subtilis 95% in the n-configuration while the free intracellular (no. 168) used in these experiments was a generous gift of Dr. alanine in both organisms after treatment with ,8-chloro- Z. -
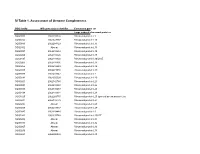
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein