Effects of Pre-Existing Orthopoxvirus-Specific Immunity On
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Munich Outbreak of Cutaneous Cowpox Infection: Transmission by Infected Pet Rats
Acta Derm Venereol 2012; 92: 126–131 INVESTIGATIVE REPORT The Munich Outbreak of Cutaneous Cowpox Infection: Transmission by Infected Pet Rats Sandra VOGEL1, Miklós SÁRDY1, Katharina GLOS2, Hans Christian KOrting1, Thomas RUZICKA1 and Andreas WOLLENBERG1 1Department of Dermatology and Allergology, Ludwig Maximilian University, Munich, and 2Department of Dermatology, Haas and Link Animal Clinic, Germering, Germany Cowpox virus infection of humans is an uncommon, another type of orthopoxvirus, from infected pet prairie potentially fatal, skin disease. It is largely confined to dogs have recently been described in the USA, making Europe, but is not found in Eire, or in the USA, Austral the medical community aware of the risk of transmission asia, or the Middle or Far East. Patients having contact of pox viruses from pets (3). with infected cows, cats, or small rodents sporadically Seven of 8 exposed patients living in the Munich contract the disease from these animals. We report here area contracted cowpox virus infection from an unusual clinical aspects of 8 patients from the Munich area who source: rats infected with cowpox virus bought from had purchased infected pet rats from a local supplier. Pet local pet shops and reputedly from the same supplier rats are a novel potential source of local outbreaks. The caused a clinically distinctive pattern of infection, which morphologically distinctive skin lesions are mostly res was mostly restricted to the patients’ neck and trunk. tricted to the patients’ necks, reflecting the infected ani We report here dermatologically relevant aspects of mals’ contact pattern. Individual lesions vaguely resem our patients in order to alert the medical community to ble orf or Milker’s nodule, but show marked surrounding the possible risk of a zoonotic orthopoxvirus outbreak erythema, firm induration and local adenopathy. -

Where Do We Stand After Decades of Studying Human Cytomegalovirus?
microorganisms Review Where do we Stand after Decades of Studying Human Cytomegalovirus? 1, 2, 1 1 Francesca Gugliesi y, Alessandra Coscia y, Gloria Griffante , Ganna Galitska , Selina Pasquero 1, Camilla Albano 1 and Matteo Biolatti 1,* 1 Laboratory of Pathogenesis of Viral Infections, Department of Public Health and Pediatric Sciences, University of Turin, 10126 Turin, Italy; [email protected] (F.G.); gloria.griff[email protected] (G.G.); [email protected] (G.G.); [email protected] (S.P.); [email protected] (C.A.) 2 Complex Structure Neonatology Unit, Department of Public Health and Pediatric Sciences, University of Turin, 10126 Turin, Italy; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Received: 19 March 2020; Accepted: 5 May 2020; Published: 8 May 2020 Abstract: Human cytomegalovirus (HCMV), a linear double-stranded DNA betaherpesvirus belonging to the family of Herpesviridae, is characterized by widespread seroprevalence, ranging between 56% and 94%, strictly dependent on the socioeconomic background of the country being considered. Typically, HCMV causes asymptomatic infection in the immunocompetent population, while in immunocompromised individuals or when transmitted vertically from the mother to the fetus it leads to systemic disease with severe complications and high mortality rate. Following primary infection, HCMV establishes a state of latency primarily in myeloid cells, from which it can be reactivated by various inflammatory stimuli. Several studies have shown that HCMV, despite being a DNA virus, is highly prone to genetic variability that strongly influences its replication and dissemination rates as well as cellular tropism. In this scenario, the few currently available drugs for the treatment of HCMV infections are characterized by high toxicity, poor oral bioavailability, and emerging resistance. -

Characterization of Host Micrornas That Respond to DNA Virus Infection in a Crustacean Tianzhi Huang, Dandan Xu and Xiaobo Zhang*
Huang et al. BMC Genomics 2012, 13:159 http://www.biomedcentral.com/1471-2164/13/159 RESEARCH ARTICLE Open Access Characterization of host microRNAs that respond to DNA virus infection in a crustacean Tianzhi Huang, Dandan Xu and Xiaobo Zhang* Abstract Background: MicroRNAs (miRNAs) are key posttranscriptional regulators of gene expression that are implicated in many processes of eukaryotic cells. It is known that the expression profiles of host miRNAs can be reshaped by viruses. However, a systematic investigation of marine invertebrate miRNAs that respond to virus infection has not yet been performed. Results: In this study, the shrimp Marsupenaeus japonicus was challenged by white spot syndrome virus (WSSV). Small RNA sequencing of WSSV-infected shrimp at different time post-infection (0, 6, 24 and 48 h) identified 63 host miRNAs, 48 of which were conserved in other animals, representing 43 distinct families. Of the identified host miRNAs, 31 were differentially expressed in response to virus infection, of which 25 were up-regulated and six down-regulated. The results were confirmed by northern blots. The TargetScan and miRanda algorithms showed that most target genes of the differentially expressed miRNAs were related to immune responses. Gene ontology analysis revealed that immune signaling pathways were mediated by these miRNAs. Evolutionary analysis showed that three of them, miR-1, miR-7 and miR-34, are highly conserved in shrimp, fruit fly and humans and function in the similar pathways. Conclusions: Our study provides the first large-scale characterization of marine invertebrate miRNAs that respond to virus infection. This will help to reveal the molecular events involved in virus-host interactions mediated by miRNAs and their evolution in animals. -

Specimen Type, Collection Methods, and Diagnostic Assays Available For
Specimen type, collection methods, and diagnostic assays available for the detection of poxviruses from human specimens by the Poxvirus and Rabies Branch, Centers for Disease Control and Prevention1. Specimen Orthopoxvirus Parapoxvirus Yatapoxvirus Molluscipoxvirus Specimen type collection method PCR6 Culture EM8 IHC9,10 Serology11 PCR12 EM8 IHC9,10 PCR13 EM8 PCR EM8 Lesion material Fresh or frozen Swab 5 Lesion material [dry or in media ] [vesicle / pustule Formalin fixed skin, scab / crust, etc.] Paraffin block Fixed slide(s) Container Lesion fluid Swab [vesicle / pustule [dry or in media5] fluid, etc.] Touch prep slide Blood EDTA2 EDTA tube 7 Spun or aliquoted Serum before shipment Spun or aliquoted Plasma before shipment CSF3,4 Sterile 1. The detection of poxviruses by electron microscopy (EM) and immunohistochemical staining (IHC) is performed by the Infectious Disease Pathology Branch of the CDC. 2. EDTA — Ethylenediaminetetraacetic acid. 3. CSF — Cerebrospinal fluid. 4. In order to accurately interpret test results generated from CSF specimens, paired serum must also be submitted. 5. If media is used to store and transport specimens a minimal amount should be used to ensure as little dilution of DNA as possible. 6. Orthopoxvirus generic real-time polymerase chain reaction (PCR) assays will amplify DNA from numerous species of virus within the Orthopoxvirus genus. Species-specific real-time PCR assays are available for selective detection of DNA from variola virus, vaccinia virus, monkeypox virus, and cowpox virus. 7. Blood is not ideal for the detection of orthopoxviruses by PCR as the period of viremia has often passed before sampling occurs. 8. EM can reveal the presence of a poxvirus in clinical specimens or from virus culture, but this technique cannot differentiate between virus species within the same genus. -
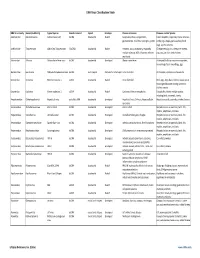
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

Monkeypox Virus Emergence in Wild Chimpanzees Reveals Distinct Clinical Outcomes and Viral Diversity
ARTICLES https://doi.org/10.1038/s41564-020-0706-0 Monkeypox virus emergence in wild chimpanzees reveals distinct clinical outcomes and viral diversity Livia V. Patrono1,6, Kamilla Pléh1,2,6, Liran Samuni2,3, Markus Ulrich1, Caroline Röthemeier1, Andreas Sachse1, Silvia Muschter4, Andreas Nitsche4, Emmanuel Couacy-Hymann5, Christophe Boesch2,3, Roman M. Wittig 2,3, Sébastien Calvignac-Spencer1 and Fabian H. Leendertz 1 ✉ Monkeypox is a viral zoonotic disease on the rise across endemic habitats. Despite the growing importance of monkeypox virus, our knowledge on its host spectrum and sylvatic maintenance is limited. Here, we describe the recent repeated emergence of monkeypox virus in a wild, human-habituated western chimpanzee (Pan troglodytes verus, hereafter chimpanzee) population from Taï National Park, Ivory Coast. Through daily monitoring, we show that further to causing its typical exanthematous syn- drome, monkeypox can present itself as a severe respiratory disease without a diffuse rash. By analysing 949 non-invasively collected samples, we identify the circulation of at least two distinct monkeypox virus lineages and document the shedding of infectious particles in faeces and flies, suggesting that they could mediate indirect transmission. We also show that the carnivo- rous component of the Taï chimpanzees’ diet, mainly consisting of the sympatric monkeys they regularly hunt, did not change nor shift towards rodent consumption (the presumed reservoir) before the outbreaks, suggesting that the sudden emergence of monkeypox virus in this population is probably due to changes in the ecology of the virus itself. Using long-term mortality surveillance data from Taï National Park, we provide evidence of little to no prior viral activity over at least two decades. -

Risk Groups: Viruses (C) 1988, American Biological Safety Association
Rev.: 1.0 Risk Groups: Viruses (c) 1988, American Biological Safety Association BL RG RG RG RG RG LCDC-96 Belgium-97 ID Name Viral group Comments BMBL-93 CDC NIH rDNA-97 EU-96 Australia-95 HP AP (Canada) Annex VIII Flaviviridae/ Flavivirus (Grp 2 Absettarov, TBE 4 4 4 implied 3 3 4 + B Arbovirus) Acute haemorrhagic taxonomy 2, Enterovirus 3 conjunctivitis virus Picornaviridae 2 + different 70 (AHC) Adenovirus 4 Adenoviridae 2 2 (incl animal) 2 2 + (human,all types) 5 Aino X-Arboviruses 6 Akabane X-Arboviruses 7 Alastrim Poxviridae Restricted 4 4, Foot-and- 8 Aphthovirus Picornaviridae 2 mouth disease + viruses 9 Araguari X-Arboviruses (feces of children 10 Astroviridae Astroviridae 2 2 + + and lambs) Avian leukosis virus 11 Viral vector/Animal retrovirus 1 3 (wild strain) + (ALV) 3, (Rous 12 Avian sarcoma virus Viral vector/Animal retrovirus 1 sarcoma virus, + RSV wild strain) 13 Baculovirus Viral vector/Animal virus 1 + Togaviridae/ Alphavirus (Grp 14 Barmah Forest 2 A Arbovirus) 15 Batama X-Arboviruses 16 Batken X-Arboviruses Togaviridae/ Alphavirus (Grp 17 Bebaru virus 2 2 2 2 + A Arbovirus) 18 Bhanja X-Arboviruses 19 Bimbo X-Arboviruses Blood-borne hepatitis 20 viruses not yet Unclassified viruses 2 implied 2 implied 3 (**)D 3 + identified 21 Bluetongue X-Arboviruses 22 Bobaya X-Arboviruses 23 Bobia X-Arboviruses Bovine 24 immunodeficiency Viral vector/Animal retrovirus 3 (wild strain) + virus (BIV) 3, Bovine Bovine leukemia 25 Viral vector/Animal retrovirus 1 lymphosarcoma + virus (BLV) virus wild strain Bovine papilloma Papovavirus/ -

Activity of the Anti-Orthopoxvirus Compound ST-246 Against Vaccinia, Cowpox and Camelpox Viruses in Cell Monolayers and Organotypic Raft Cultures
Andrei 21/11/07 15:46 Page 1205 Antiviral Therapy 12:1205–1216 Activity of the anti-orthopoxvirus compound ST-246 against vaccinia, cowpox and camelpox viruses in cell monolayers and organotypic raft cultures Sophie Duraffour1,2, Robert Snoeck1, Rita de Vos 3, Joost J van Den Oord3, Jean-Marc Crance 2, Daniel Garin2, Dennis E Hruby4, Robert Jordan4, Erik De Clercq1 and Graciela Andrei1* 1Rega Institute For Medical Research, KU Leuven, Leuven, Belgium 2CRSSA Emile Pardé, Virology Laboratory, La Tronche, France 3Pathology Department, UZ Leuven, Leuven, Belgium 4SIGA Technologies, Inc., Corvallis, Oregon, CA, USA *Corresponding author: Tel: +32 16 33 73 72; Fax: +32 16 33 73 40; E-mail: [email protected] Background: The potential use of variola virus as a biolog- Results: ST-246 inhibited preferentially the production of ical weapon has renewed efforts in the development of extracellular virus compared with intracellular virus antiviral agents against orthopoxviruses. ST-246 [4- production in HEL and PHK cells (for VV) and in PHK cells trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-di (for CMLV). In organotypic epithelial raft cultures, ST-246 oxo-4,6-ethenocycloprop [f]isoindol-2(1H)-yl)-benza- at 20 μg/ml inhibited extracellular VV and CMLV produc- mide] is an anti-orthopoxvirus compound active against tion by 6 logs, whereas intracellular virus yield was several orthopoxviruses including vaccinia virus (VV), reduced by 2 logs. In the case of CPV, both extracellular cowpox virus (CPV), camelpox virus (CMLV), ectromelia and intracellular virus production were completely inhib- virus (ECTV) and variola virus in cell culture. -

Introduction to Viroids and Prions
Harriet Wilson, Lecture Notes Bio. Sci. 4 - Microbiology Sierra College Introduction to Viroids and Prions Viroids – Viroids are plant pathogens made up of short, circular, single-stranded RNA molecules (usually around 246-375 bases in length) that are not surrounded by a protein coat. They have internal base-pairs that cause the formation of folded, three-dimensional, rod-like shapes. Viroids apparently do not code for any polypeptides (proteins), but do cause a variety of disease symptoms in plants. The mechanism for viroid replication is not thoroughly understood, but is apparently dependent on plant enzymes. Some evidence suggests they are related to introns, and that they may also infect animals. Disease processes may involve RNA-interference or activities similar to those involving mi-RNA. Prions – Prions are proteinaceous infectious particles, associated with a number of disease conditions such as Scrapie in sheep, Bovine Spongiform Encephalopathy (BSE) or Mad Cow Disease in cattle, Chronic Wasting Disease (CWD) in wild ungulates such as muledeer and elk, and diseases in humans including Creutzfeld-Jacob disease (CJD), Gerstmann-Straussler-Scheinker syndrome (GSS), Alpers syndrome (in infants), Fatal Familial Insomnia (FFI) and Kuru. These diseases are characterized by loss of motor control, dementia, paralysis, wasting and eventually death. Prions can be transmitted through ingestion, tissue transplantation, and through the use of comtaminated surgical instruments, but can also be transmitted from one generation to the next genetically. This is because prion proteins are encoded by genes normally existing within the brain cells of various animals. Disease is caused by the conversion of normal cell proteins (glycoproteins) into prion proteins. -

Here, There, and Everywhere: the Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses
viruses Review Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses Natalia Ingrid Oliveira Silva, Jaqueline Silva de Oliveira, Erna Geessien Kroon , Giliane de Souza Trindade and Betânia Paiva Drumond * Laboratório de Vírus, Departamento de Microbiologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais: Belo Horizonte, Minas Gerais 31270-901, Brazil; [email protected] (N.I.O.S.); [email protected] (J.S.d.O.); [email protected] (E.G.K.); [email protected] (G.d.S.T.) * Correspondence: [email protected] Abstract: The global emergence of zoonotic viruses, including poxviruses, poses one of the greatest threats to human and animal health. Forty years after the eradication of smallpox, emerging zoonotic orthopoxviruses, such as monkeypox, cowpox, and vaccinia viruses continue to infect humans as well as wild and domestic animals. Currently, the geographical distribution of poxviruses in a broad range of hosts worldwide raises concerns regarding the possibility of outbreaks or viral dissemination to new geographical regions. Here, we review the global host ranges and current epidemiological understanding of zoonotic orthopoxviruses while focusing on orthopoxviruses with epidemic potential, including monkeypox, cowpox, and vaccinia viruses. Keywords: Orthopoxvirus; Poxviridae; zoonosis; Monkeypox virus; Cowpox virus; Vaccinia virus; host range; wild and domestic animals; emergent viruses; outbreak Citation: Silva, N.I.O.; de Oliveira, J.S.; Kroon, E.G.; Trindade, G.d.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range 1. Poxvirus and Emerging Diseases and Geographic Distribution of Zoonotic diseases, defined as diseases or infections that are naturally transmissible Zoonotic Orthopoxviruses. Viruses from vertebrate animals to humans, represent a significant threat to global health [1]. -

Substantial Equivalence Determination Decision Summary
510(k) SUBSTANTIAL EQUIVALENCE DETERMINATION DECISION SUMMARY A. 510(k) Number: K181205 B. Purpose for Submission: Modification of device C. Measurand: Non-variola Orthopoxvirus DNA target sequence D. Type of Test: In vitro molecular diagnostic test E. Applicant: Centers for Disease Control and Prevention F. Proprietary and Established Names: Non-variola Orthopoxvirus Real-time PCR Primer and Probe Set G. Regulatory Information: 1. Regulation section: 21 CFR 866.3315: Nucleic acid based reagents for detection of non-variola orthopoxviruses 2. Classification: Class II (Special Controls) 3. Product code: PBK 4. Panel: Microbiology (83) 1 H. Intended Use: 1. Intended use(s): The Non-variola Orthopoxvirus Real-time PCR Primer and Probe Set is intended for the in vitro qualitative presumptive detection of non-variola Orthopoxvirus DNA extracted from human pustular or vesicular rash specimens and viral cell culture lysates submitted to a Laboratory Response Network (LRN) reference laboratory. The assay detects non- variola Orthopoxvirus DNA, including Vaccinia, Cowpox, Monkeypox and Ectromelia viruses at varying concentrations. This assay does not differentiate Vaccinia virus or Monkeypox virus from other Orthopoxviruses detected by this assay and does not detect Variola virus. Refer to the CDC algorithm, Acute, Generalized Vesicular or Pustular Rash Illness Testing Protocol in the United States for recommended testing and evaluation algorithms for patients presenting with acute, generalized pustular or vesicular rash illness. Results of this assay are for the presumptive identification of non-variola Orthopoxvirus DNA. These results must be used in conjunction with other diagnostic assays and clinical observations to diagnose Orthopoxvirus infection. The assay should only be used to test specimens with low/moderate risk of smallpox. -

Cross-Neutralizing and Protective Human Antibody Specificities To
Article Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections Graphical Abstract Authors Iuliia Gilchuk, Pavlo Gilchuk, Gopal Sapparapu, ..., Gary H. Cohen, Sebastian Joyce, James E. Crowe, Jr. Correspondence [email protected] In Brief Protective immunity against smallpox and other members of the orthopoxvirus family requires the cooperation of antibodies that target different viral proteins at distinct stages of maturation of the virus. Highlights d Orthopoxviruses elicit a complex B cell immune response reactive to diverse antigens d A large fraction of orthopoxvirus neutralizing mAbs possess cross-neutralizing activity d Six principal mAb specificities participate in cross- neutralization and protection d Most efficient protection is achieved by mixture of diverse mAbs specificities Gilchuk et al., 2016, Cell 167, 684–694 October 20, 2016 ª 2016 Elsevier Inc. http://dx.doi.org/10.1016/j.cell.2016.09.049 Article Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections Iuliia Gilchuk,1 Pavlo Gilchuk,2,3 Gopal Sapparapu,1,4 Rebecca Lampley,1 Vidisha Singh,1 Nurgun Kose,1 David L. Blum,1 Laura J. Hughes,5 Panayampalli S. Satheshkumar,5 Michael B. Townsend,5 Ashley V. Kondas,5 Zachary Reed,5,6 Zachary Weiner,6 Victoria A. Olson,5 Erika Hammarlund,7 Hans-Peter Raue,7 Mark K. Slifka,7 James C. Slaughter,8 Barney S. Graham,9 Kathryn M. Edwards,4 Roselyn J. Eisenberg,10 Gary H. Cohen,11 Sebastian Joyce,2,3 and James E. Crowe, Jr.1,2,4,12,* 1The Vanderbilt Vaccine Center, Vanderbilt