Gibbs Paradox of Entropy of Mixing: Experimental Facts, Its Rejection and the Theoretical Consequences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Gibbs' Paradox and the Definition of Entropy
Entropy 2008, 10, 15-18 entropy ISSN 1099-4300 °c 2008 by MDPI www.mdpi.org/entropy/ Full Paper Gibbs’ Paradox and the Definition of Entropy Robert H. Swendsen Physics Department, Carnegie Mellon University, Pittsburgh, PA 15213, USA E-Mail: [email protected] Received: 10 December 2007 / Accepted: 14 March 2008 / Published: 20 March 2008 Abstract: Gibbs’ Paradox is shown to arise from an incorrect traditional definition of the entropy that has unfortunately become entrenched in physics textbooks. Among its flaws, the traditional definition predicts a violation of the second law of thermodynamics when applied to colloids. By adopting Boltzmann’s definition of the entropy, the violation of the second law is eliminated, the properties of colloids are correctly predicted, and Gibbs’ Paradox vanishes. Keywords: Gibbs’ Paradox, entropy, extensivity, Boltzmann. 1. Introduction Gibbs’ Paradox [1–3] is based on a traditional definition of the entropy in statistical mechanics found in most textbooks [4–6]. According to this definition, the entropy of a classical system is given by the product of Boltzmann’s constant, k, with the logarithm of a volume in phase space. This is shown in many textbooks to lead to the following equation for the entropy of a classical ideal gas of distinguishable particles, 3 E S (E; V; N) = kN[ ln V + ln + X]; (1) trad 2 N where X is a constant. Most versions of Gibbs’ Paradox, including the one I give in this section, rest on the fact that Eq. 1 is not extensive. There is another version of Gibbs’ Paradox that involves the mixing of two gases, which I will discuss in the third section of this paper. -

Calculating the Configurational Entropy of a Landscape Mosaic
Landscape Ecol (2016) 31:481–489 DOI 10.1007/s10980-015-0305-2 PERSPECTIVE Calculating the configurational entropy of a landscape mosaic Samuel A. Cushman Received: 15 August 2014 / Accepted: 29 October 2015 / Published online: 7 November 2015 Ó Springer Science+Business Media Dordrecht (outside the USA) 2015 Abstract of classes and proportionality can be arranged (mi- Background Applications of entropy and the second crostates) that produce the observed amount of total law of thermodynamics in landscape ecology are rare edge (macrostate). and poorly developed. This is a fundamental limitation given the centrally important role the second law plays Keywords Entropy Á Landscape Á Configuration Á in all physical and biological processes. A critical first Composition Á Thermodynamics step to exploring the utility of thermodynamics in landscape ecology is to define the configurational entropy of a landscape mosaic. In this paper I attempt to link landscape ecology to the second law of Introduction thermodynamics and the entropy concept by showing how the configurational entropy of a landscape mosaic Entropy and the second law of thermodynamics are may be calculated. central organizing principles of nature, but are poorly Result I begin by drawing parallels between the developed and integrated in the landscape ecology configuration of a categorical landscape mosaic and literature (but see Li 2000, 2002; Vranken et al. 2014). the mixing of ideal gases. I propose that the idea of the Descriptions of landscape patterns, processes of thermodynamic microstate can be expressed as unique landscape change, and propagation of pattern-process configurations of a landscape mosaic, and posit that relationships across scale and through time are all the landscape metric Total Edge length is an effective governed and constrained by the second law of measure of configuration for purposes of calculating thermodynamics (Cushman 2015). -

Gibbs Paradox: Mixing and Non Mixing Potentials T
Physics Education 1 Jul - Sep 2016 Gibbs paradox: Mixing and non mixing potentials T. P. Suresh 1∗, Lisha Damodaran 1 and K. M. Udayanandan 2 1 School of Pure and Applied Physics Kannur University, Kerala- 673 635, INDIA *[email protected] 2 Nehru Arts and Science College, Kanhangad Kerala- 671 314, INDIA (Submitted 12-03-2016, Revised 13-05-2016) Abstract Entropy of mixing leading to Gibbs paradox is studied for different physical systems with and without potentials. The effect of potentials on the mixing and non mixing character of physical systems is discussed. We hope this article will encourage students to search for new problems which will help them understand statistical mechanics better. 1 Introduction namics of any system (which is the main aim of statistical mechanics) we need to calculate Statistical mechanics is the study of macro- the canonical partition function, and then ob- scopic properties of a system from its mi- tain macro properties like internal energy, en- croscopic description. In the ensemble for- tropy, chemical potential, specific heat, etc. malism introduced by Gibbs[1] there are of the system from the partition function. In three ensembles-micro canonical, canonical this article we make use of the canonical en- and grand canonical ensemble. Since we are semble formalism to study the extensive char- not interested in discussing quantum statis- acter of entropy and then to calculate the tics we will use the canonical ensemble for in- entropy of mixing. It is then used to ex- troducing our ideas. To study the thermody- plain the Gibbs paradox. Most text books Volume 32, Number 3, Article Number: 4. -

Gibb's Paradox for Distinguishable
Gibb’s paradox for distinguishable K. M. Udayanandan1, R. K. Sathish1, A. Augustine2 1Department of Physics, Nehru Arts and Science College, Kerala-671 314, India. 2Department of Physics, University of Kannur, Kerala 673 635, India. E-mail: [email protected] (Received 22 March 2014, accepted 17 November 2014) Abstract Boltzmann Correction Factor (BCF) N! is used in micro canonical ensemble and canonical ensemble as a dividing term to avoid Gibbs paradox while finding the number of states and partition function for ideal gas. For harmonic oscillators this factor does not come since they are considered to be distinguishable. We here show that BCF comes twice for harmonic oscillators in grand canonical ensemble for entropy to be extensive in classical statistics. Then we extent this observation for all distinguishable systems. Keywords: Gibbs paradox, harmonic oscillator, distinguishable particles. Resumen El factor de corrección de Boltzmann (BCF) N! se utiliza en ensamble microcanónico y ensamble canónico como un término divisor para evitar la paradoja de Gibbs, mientras se busca el número de estados y la función de partición para un gas ideal. Para osciladores armónicos este factor no viene sin que éstos sean considerados para ser distinguibles. Mostramos aquí que BCF llega dos veces para osciladores armónicos en ensamble gran canónico para que la entropía sea ampliamente usada en la estadística clásica. Luego extendemos esta observación para todos los sistemas distinguibles. Palabras clave: paradoja de Gibbs, oscilador armónico, partículas distinguibles. PACS: 03.65.Ge, 05.20-y ISSN 1870-9095 I. INTRODUCTION Statistical mechanics is a mathematical formalism by which II. CANONICAL ENSEMBLE one can determine the macroscopic properties of a system from the information about its microscopic states. -

Classical Particle Indistinguishability, Precisely
Classical Particle Indistinguishability, Precisely James Wills∗1 1Philosophy, Logic and Scientific Method, London School of Economics Forthcoming in The British Journal for the Philosophy of Science Abstract I present a new perspective on the meaning of indistinguishability of classical particles. This leads to a solution to the problem in statisti- cal mechanics of justifying the inclusion of a factor N! in a probability distribution over the phase space of N indistinguishable classical par- ticles. 1 Introduction Considerations of the identity of objects have long been part of philosophical discussion in the natural sciences and logic. These considerations became particularly pertinent throughout the twentieth century with the develop- ment of quantum physics, widely recognized as having interesting and far- reaching implications concerning the identity, individuality, indiscernibility, indistinguishability1 of the elementary components of our ontology. This discussion continues in both the physics and philosophy literature2. ∗[email protected] 1. This menagerie of terms in the literature is apt to cause confusion, especially as there is no clear consensus on what exactly each of these terms mean. Throughout the rest of the paper, I will be concerned with giving a precise definition of a concept which I will label `indistinguishability', and which, I think, matches with how most other commentators use the word. But I believe it to be a distinct debate whether particles are `identical', `individual', or `indiscernible'. The literature I address in this paper therefore does not, for example, overlap with the debate over the status of Leibniz's PII. 2. See French (2000) for an introduction to this discussion and a comprehensive list of references. -

Chemistry C3102-2006: Polymers Section Dr. Edie Sevick, Research School of Chemistry, ANU 5.0 Thermodynamics of Polymer Solution
Chemistry C3102-2006: Polymers Section Dr. Edie Sevick, Research School of Chemistry, ANU 5.0 Thermodynamics of Polymer Solutions In this section, we investigate the solubility of polymers in small molecule solvents. Solubility, whether a chain goes “into solution”, i.e. is dissolved in solvent, is an important property. Full solubility is advantageous in processing of polymers; but it is also important for polymers to be fully insoluble - think of plastic shoe soles on a rainy day! So far, we have briefly touched upon thermodynamic solubility of a single chain- a “good” solvent swells a chain, or mixes with the monomers, while a“poor” solvent “de-mixes” the chain, causing it to collapse upon itself. Whether two components mix to form a homogeneous solution or not is determined by minimisation of a free energy. Here we will express free energy in terms of canonical variables T,P,N , i.e., temperature, pressure, and number (of moles) of molecules. The free energy { } expressed in these variables is the Gibbs free energy G G(T,P,N). (1) ≡ In previous sections, we referred to the Helmholtz free energy, F , the free energy in terms of the variables T,V,N . Let ∆Gm denote the free eneregy change upon homogeneous mix- { } ing. For a 2-component system, i.e. a solute-solvent system, this is simply the difference in the free energies of the solute-solvent mixture and pure quantities of solute and solvent: ∆Gm G(T,P,N , N ) (G0(T,P,N )+ G0(T,P,N )), where the superscript 0 denotes the ≡ 1 2 − 1 2 pure component. -

Lecture 6: Entropy
Matthew Schwartz Statistical Mechanics, Spring 2019 Lecture 6: Entropy 1 Introduction In this lecture, we discuss many ways to think about entropy. The most important and most famous property of entropy is that it never decreases Stot > 0 (1) Here, Stot means the change in entropy of a system plus the change in entropy of the surroundings. This is the second law of thermodynamics that we met in the previous lecture. There's a great quote from Sir Arthur Eddington from 1927 summarizing the importance of the second law: If someone points out to you that your pet theory of the universe is in disagreement with Maxwell's equationsthen so much the worse for Maxwell's equations. If it is found to be contradicted by observationwell these experimentalists do bungle things sometimes. But if your theory is found to be against the second law of ther- modynamics I can give you no hope; there is nothing for it but to collapse in deepest humiliation. Another possibly relevant quote, from the introduction to the statistical mechanics book by David Goodstein: Ludwig Boltzmann who spent much of his life studying statistical mechanics, died in 1906, by his own hand. Paul Ehrenfest, carrying on the work, died similarly in 1933. Now it is our turn to study statistical mechanics. There are many ways to dene entropy. All of them are equivalent, although it can be hard to see. In this lecture we will compare and contrast dierent denitions, building up intuition for how to think about entropy in dierent contexts. The original denition of entropy, due to Clausius, was thermodynamic. -

Solutions Mole Fraction of Component a = Xa Mass Fraction of Component a = Ma Volume Fraction of Component a = Φa Typically We
Solutions Mole fraction of component A = xA Mass Fraction of component A = mA Volume Fraction of component A = fA Typically we make a binary blend, A + B, with mass fraction, m A, and want volume fraction, fA, or mole fraction , xA. fA = (mA/rA)/((mA/rA) + (m B/rB)) xA = (mA/MWA)/((mA/MWA) + (m B/MWB)) 1 Solutions Three ways to get entropy and free energy of mixing A) Isothermal free energy expression, pressure expression B) Isothermal volume expansion approach, volume expression C) From statistical thermodynamics 2 Mix two ideal gasses, A and B p = pA + pB pA is the partial pressure pA = xAp For single component molar G = µ µ 0 is at p0,A = 1 bar At pressure pA for a pure component µ A = µ0,A + RT ln(p/p0,A) = µ0,A + RT ln(p) For a mixture of A and B with a total pressure ptot = p0,A = 1 bar and pA = xA ptot For component A in a binary mixture µ A(xA) = µ0.A + RT xAln (xA ptot/p0,A) = µ0.A + xART ln (xA) Notice that xA must be less than or equal to 1, so ln xA must be negative or 0 So the chemical potential has to drop in the solution for a solution to exist. Ideal gasses only have entropy so entropy drives mixing in this case. This can be written, xA = exp((µ A(xA) - µ 0.A)/RT) Which indicates that xA is the Boltzmann probability of finding A 3 Mix two real gasses, A and B µ A* = µ 0.A if p = 1 4 Ideal Gas Mixing For isothermal DU = CV dT = 0 Q = W = -pdV For ideal gas Q = W = nRTln(V f/Vi) Q = DS/T DS = nRln(V f/Vi) Consider a process of expansion of a gas from VA to V tot The change in entropy is DSA = nARln(V tot/VA) = - nARln(VA/V tot) Consider an isochoric mixing process of ideal gasses A and B. -
PHYS 521: Statistical Mechanics Homework #4
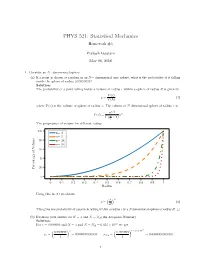
PHYS 521: Statistical Mechanics Homework #4 Prakash Gautam May 08, 2018 1. Consider an N−dimensional sphere. (a) If a point is chosen at random in an N− dimensional unit sphere, what is the probability of it falling inside the sphere of radius 0:99999999? Solution: The probability of a point falling inside a volume of radius r within a sphere of radius R is given by V (r) p = (1) V (R) where V (x) is the volume of sphere of radius x. The volume of N dimensional sphere of radius x is n=2 (π ) n V (x) = n x Γ 2 + 1 The progression of volume for different radius. 100 n= 2 n= 5 80 n= 24 n= 54 60 40 20 Percentage of Volume 0 0 0:1 0:2 0:3 0:4 0:5 0:6 0:7 0:8 0:9 1 Radius Using this in (1) we obtain ( ) r n p = (2) R This gives the probability of a particle falling within a radius r in a Ndimensional sphere of radius R. □ (b) Evaluate your answer for N = 3 and N = NA(the Avogadro Number) Solution: 23 For r = 0:999999 and N = 3 and N = NA = 6:023 × 10 we get ( ) ( ) × 23 0:999999 3 0:999999 6:023 10 p = = 0:999997000003 p = = 0:0000000000000 3 1 NA 1 1 The probability of a particle falling within the radius nearly 1 in higher two-dimensional sphere is vanishningly small. □ (c) What do these results say about the equivalence of the definitions of entropy in terms of either of the total phase space volume of the volume of outermost energy shell? Solution: Considering a phase space volume bounded by E + ∆ where ∆ ≪ E. -

A Note on Gibbs Paradox 1. Introduction
1 A note on Gibbs paradox P. Radhakrishnamurty 282, Duo Marvel Layout, Anathapuragate, Yelahanka, Bangalore 560064, India. email: [email protected] Abstract We show in this note that Gibbs paradox arises not due to application of thermodynamic principles, whether classical or statistical or even quantum mechanical, but due to incorrect application of mathematics to the process of mixing of ideal gases. ------------------------------------------------------------------------------------------------------------------------------- Key Words: Gibbs paradox, Amagat’s law, Dalton’s law, Ideal gas mixtures, entropy change 1. Introduction ‘It has always been believed that Gibbs paradox embodied profound thought’, Schrodinger [1]. It is perhaps such a belief that underlies the continuing discussion of Gibbs paradox over the last hundred years or more. Literature available on Gibbs paradox is vast [1-15]. Lin [16] and Cheng [12] give extensive list of some of the important references on the issue, besides their own contributions. Briefly stated, Gibbs paradox arises in the context of application of the statistical mechanical analysis to the process of mixing of two ideal gas samples. The discordant values of entropy change associated with the process, in the two cases: 1. when the two gas samples contain the same species and, 2. when they contain two different species constitutes the gist of Gibbs paradox. A perusal of the history of the paradox shows us that: Boltzmann, through the application of statistical mechanics to the motion of particles (molecules) and the distribution of energy among the particles contained in a system, developed the equation: S = log W + S(0), connecting entropy S of the system to the probability W of a microscopic state corresponding to a given macroscopic equilibrium state of the system, S(0) being the entropy at 0K. -

Chemistry 163B Absolute Entropies and Entropy of Mixing
Chemistry 163B Winter 2012 Handouts for Third Law and Entropy of Mixing (ideal gas, distinguishable molecules) Chemistry 163B Absolute Entropies and Entropy of Mixing 1 APPENDIX A: Hf, Gf, BUT S (no Δ, no “sub f ”) Hºf Gºf Sº 2 1 Chemistry 163B Winter 2012 Handouts for Third Law and Entropy of Mixing (ideal gas, distinguishable molecules) Third Law of Thermodynamics The entropy of any perfect crystalline substance approaches 0 as Tô 0K S=k ln W for perfectly ordered crystalline substance W ô 1 as Tô 0K S ô 0 3 to calculate absolute entropy from measurements (E&R pp. 101-103, Figs 5.8-5.10) T2 CP S ABCDE,,,, dT ABCDE,,,, T T1 H S T ABCD E S S S S III II III I g 0 + + + + S298.15 SK(0 ) SA S SB S 0 23.66 III II 23.66 43.76 II I at 23.66K at4 3.76K + + + SC + S SD + S SE 43.7654 .39 I 54.3990.20 g 90.30298.15 at54 .39K at90.20 K 4 2 Chemistry 163B Winter 2012 Handouts for Third Law and Entropy of Mixing (ideal gas, distinguishable molecules) full calculation of Sº298 for O2 (g) (Example Problem 5.9, E&R pp103-104 [96-97]2nd) SJ K11 mol S(0K ) 0 SA 0 23.66 8.182 S III II at 23.66K 3.964 SB 23.66 43.76 19.61 S II I at 43.76K 16.98 SC 43.76 54.39 10.13 S I at 54.39K 8.181 SD 54.39 90.20 27.06 Sg at 90.20K 75.59 SE 90.20 298.15 35.27 total 204.9 J K-1 mol-1 5 Sreaction from absolute entropies nAA + nBB ônCC + nDD at 298K 0000 SnSnSnSnS298 298 298 298 reaction CCDAB D A B SK00()298 S reaction i 298 i i S 0 are 3rd Law entropies (e.g.