Genetic Analysis of Tarantulas in the Genus Brachypelma Using Inter Simple Sequence Repeats (ISSR)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

BMB-WRC Animal Inventory
Department of Environment and Natural Resources BIODIVERSITY MANAGEMENT BUREAU Quezon Avenue, Diliman, Quezon City INVENTORY OF LIVE ANIMALS AT THE BMB-WILDLIFE RESCUE CENTER AS OF JULY 31, 2020 SPECIES STOCK ON HAND (AS OF COMMON NAME SCIENTIFIC NAME JULY 31, 2020) MAMMALS ENDEMIC / INDIGENOUS 1. Northern luzon cloud Ploeomys pallidus 1 rat 2. Palawan bearcat Arctictis binturong 2 3. Philippine deer Rusa marianna 2 4. Philippine monkey or Macaca fascicularis 92 Long-tailed macaque 5. Philippine palm civet Paradoxurus hermaphroditus 6 EXOTIC 6. Hedgehog Atelerix frontalis 1 7. Serval cat Leptailurus serval 2 8. Sugar glider Petaurus breviceps 58 9. Tiger Panthera tigris 2 10. Vervet monkey Chlorocebus pygerythrus 1 11. White handed gibbon Hylobates lar 1 Sub-total A 168 (Mammals) AVIANS ENDEMIC / INDIGENOUS 12. Black kite Milvus migrans 1 13. Black-crowned night Nycticorax nycticorax 1 heron 14. Blue-naped parrot Tanygnathus lucionensis 4 15. Brahminy kite Haliastur indus 41 16. Changeable hawk Spizaetus cirrhatus 6 eagle 17. Crested goshawk Accipiter trivirgatus 1 18. Crested serpent eagle Spilornis cheela 24 19. Green imperial pigeon Ducula aenea 2 20. Grey-headed fish eagle Haliaeetus ichthyaetus 1 21. Nicobar pigeon Caloenas nicobarica 1 22. Palawan hornbill Anthracoceros marchei 2 23. Palawan talking myna Gracula religiosa 3 24. Philippine eagle Pithecophaga jefferyi 1 25. Philippine hanging Loriculus philippensis 11 parrot 26. Philippine hawk eagle Spizaetus philippensis 12 27. Philippine horned Bubo philippensis 9 (eagle) owl 28. Philippine Scops owl Otus megalotis 5 29. Pink-necked pigeon Treron vernans 1 30. Pinsker's hawk eagle Spizaetus pinskerii 1 31. Red turtle dove Streptopelia tranquebarica 1 32. -

A New Species of North American Tarantula , Aphonopelma Paloma (Araneae, Mygalomorphae, Theraphosidae )
1992. The Journal of Arachnology 20 :189—199 A NEW SPECIES OF NORTH AMERICAN TARANTULA , APHONOPELMA PALOMA (ARANEAE, MYGALOMORPHAE, THERAPHOSIDAE ) Thomas R. Prentice: Department of Entomology, University of California, Riverside , California 92521, US A ABSTRACT. Aphonopelma paloma new species, is distinguished from all other North American tarantula s by its unusually small size and presence of setae partially or completely dividing the scopula of tarsus IV i n both sexes. Both sexes also are characterized by a general reduction of the scopula on metatarsus IV . Males are characterized by a swollen third femur . In 1939 and 1940 R. V. Chamberlin and W. with anterior and posterior edges in the same Ivie described almost all of the currently recog- plane. All ink drawings except femora were aide d nized North American theraphosid spiders . De- by a camera lucida. Palpal bulb and seminal re- spite the acknowledged significance of their work , ceptacles were cleared in 10% NaOH (for 12 hr. it is difficult to apply Chamberlin's keys wit h at 50 °C.) prior to illustration . Scanning electron much success even in dealing with specimen s micrographs were taken with a JEOL JSM C35 . from type localities, primarily because their small Abbreviations for eyes are standard for Araneae. sample sizes did not allow variational assess- For leg spination, abbreviations are as follows : ment. Eleven of these species descriptions were a = apical, b = basal, d = dorsal, e = preapical, based on single males, five on single females, an d L = left, m = medial, p = prolateral direction, r three on two males each (Chamberlin & Ivie 1939; = retrolateral direction, R = right, usu . -

Redescription of the Holotypes of Mygalarachnae Ausserer 1871 And
ARTÍCULO: Redescription of the holotypes of Mygalarachnae Ausserer 1871 and Harpaxictis Simon (1892) (Araneae: Theraphosidae) with rebuttal of their synonymy with Sericopelma Ausserer 1875. Ray Gabriel and Stuart.J. Longhorn ARTÍCULO: Abstract: Redescription of the holotypes of The examination of specimens from various Neotropical tarantula genera (The- Mygalarachnae Ausserer 1871 and raphosidae) indicated the unique nature of monotypic genera Mygalarachnae Harpaxictis Simon (1892) (Araneae: Ausserer 1871 and Harpaxictis Simon 1892. Review of the both holotypes leads Theraphosidae) with rebuttal of their us to argue that Mygalarachnae and Harpaxictis should be removed from their synonymy with Sericopelma current synonymy with Sericopelma Ausserer 1875. The presence of type I and Ausserer 1875. III urticating hairs on the holotype specimen of Mygalarachnae firmly place it in the subfamily Theraphosinae and we argue should be restored as a valid genus, Ray Gabriel so that current placement of “incertae sedis” is inappropriate. The identity of Hope Entomological Collections, Harpaxictis striatus is less certain, but here removed from synonymy with Seri- Oxford University Museum of Natural copelma, and due to a lack of other diagnostic features is suggested as nomen History, Parks Road, Oxford, dubium. Possible affinities of Mygalarachne with other valid genera of Thera- Oxon, England, OX1 3PW, UK. phosinae are briefly discussed. Key words: Sericopelma, Tarantula. Mygalarachne brevipes, Harpaxictis striatus, Stuart.J. Longhorn Taxonomy: Mygalarachne brevipes comb. rev., Harpaxictis striatus comb. rev. Dept. of Entomology. The Natural History Museum, Cromwell Road, London, SW7 5BD, Redescripción de los holotipos de Mygalarachnae Ausserer 1871 UK. y Harpaxictis Simon (1892) rechazando su sinonímia con Dept. of Biology. -

Spider and Scorpion Case
Spider and Scorpion case Black widow spider (Lactrodectus hesperus) Black widows are notorious spiders identified by the colored, hourglass-shaped mark on their abdomens. Several species answer to the name, and they are found in temperate regions around the world. This spider's bite is much feared because its venom is reported to be 15 times stronger than a rattlesnake's. In humans, bites produce muscle aches, nausea, and a paralysis of the diaphragm that can make breathing difficult; however, contrary to popular belief, most people who are bitten suffer no serious damage—let alone death. But bites can be fatal—usually to small children, the elderly, or the infirm. Fortunately, fatalities are fairly rare; the spiders are nonaggressive and bite only in self-defense, such as when someone accidentally sits on them. These spiders spin large webs in which females suspend a cocoon with hundreds of eggs. Spiderlings disperse soon after they leave their eggs, but the web remains. Black widow spiders also use their webs to ensnare their prey, which consists of flies, mosquitoes, grasshoppers, beetles, and caterpillars. Black widows are comb- footed spiders, which means they have bristles on their hind legs that they use to cover their prey with silk once it has been trapped. To feed, black widows puncture their insect prey with their fangs and administer digestive enzymes to the corpses. By using these enzymes, and their gnashing fangs, the spiders liquefy their prey's bodies and suck up the resulting fluid. Giant desert hairy scorpion (Hadrurus arizonensis) Hadrurus arizonensis is distributed throughout the Sonora and Mojave deserts. -
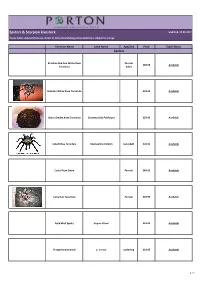
Spiders & Scorpion Livestock
Spiders & Scorpion Livestock Updated: 22.09.2017 Please Note: Livestock lists are correct at time of publishing and availability is subject to change Common Name Latin Name Age/Size Price Stock Status Spiders Brazilian Red And White Knee Female £49.95 Available Tarantula Adult Brazilian White Knee Tarantula £34.95 Available Chaco Golden Knee Tarantula Grammostola Pulchripes £29.95 Available Cobalt Blue Tarantula Haplopelma Lividum Sub Adult £49.95 Available Costa Rican Zebra Female £49.95 Available Curly Hair Tarantula Female £29.95 Available Field Wolf Spider Hogna Miami £19.95 Available Fringed Ornamental p. ornata spiderling £24.95 Available 1 / 4 Common Name Latin Name Age/Size Price Stock Status Spiders Golden Baboon Tarantula Augacephalus ezendami £39.95 Available Gooty Ornamental p. metallica spiderling £49.95 Available Green Bottle Blue Tarantula Female £49.95 Available Green Femur Birdeater Phormictopus"Green Femur" £39.95 Available Indian Ornamental Tarantula Poecilotheria regalis 2cm £24.95 Available King Baboon Tarantula Pelinobius Muticus £24.95 Available Martinique Pink Toe Tarantula 2cm £16.95 Available Mexican Fire Leg Tarantula Brachypelma Bohemi £39.95 Available Mexican Red Knee Tarantula Brachypelma Smithi £34.95 Available 2 / 4 Common Name Latin Name Age/Size Price Stock Status Spiders Mexican Red Leg Tarantula Brachypelma Emilia £39.95 Available £24.95 Available Mexican Red Rump Brachypelma Vagans Female Sub Adult Available £49.95 spiderling £19.95 Available OBT Tarantula Female Sub Adult Available £39.95 Purple Earth -

Description of Two New Species of Plesiopelma (Araneae, Theraphosidae, Theraphosinae) from Argentina
374 Ferretti & Barneche Description of two new species of Plesiopelma (Araneae, Theraphosidae, Theraphosinae) from Argentina Nelson Ferretti & Jorge Barneche Centro de Estudios Parasitológicos y de Vectores CEPAVE (CCT- CONICET- La Plata) (UNLP), Calle 2 n°584, La Plata, Argentina. ([email protected]; [email protected]) ABSTRACT. Two new species of Plesiopelma Pocock, 1901 from northern Argentina are described and diagnosed based on males and habitat descriptions are presented. Males of Plesiopelma paganoi sp. nov. differ from most of species by the absence of spiniform setae on the retrolateral face of cymbium, aspect of the palpal bulb. Plesiopelma aspidosperma sp. nov. differs from most species of the genus by the presence of spiniform setae on the retrolateral face of cymbium and it can be distinguished from P. myodes Pocock, 1901, P. longisternale (Schiapelli & Gerschman, 1942) and P. rectimanum (Mello-Leitão, 1923) by the separated palpal bulb keels and basal nodule of metatarsus I very developed. It differs from P. minense (Mello-Leitão, 1943) by the shape of the palpal bulb and basal nodule on metatarsus I well developed. Specimens were captured in Salta province, Argentina, inhabiting high cloud forests of Yungas eco-region. KEYWORDS. Taxonomy, spiders, natural history, Neotropical, Yungas. RESUMEN. Descripción de dos nuevas especies de Plesiopelma (Araneae, Theraphosidae, Theraphosinae) de Argentina. Dos nuevas especies de Plesiopelma Pocock, 1901 del norte de Argentina son diferenciadas y se describen en base a ejemplares machos y se presentan descripciones de los ambientes. Machos de Plesiopelma paganoi sp. nov. difieren de la mayoría de las especies por la ausencia de setas espiniformes en la cara retrolateral del cymbium, por la forma del órgano palpar. -

Colorado Tarantulas
Colorado Arachnids of Interest Colorado Tarantulas Scientific Name: Aphonopelma spp. (3-5 species) Class: Arachnida (Arachnids) Order: Araneae (Spiders) Family: Theraphosidae (Tarantulas) Figure 1. Male Oklahoma brown tarantula (Aphonopelma hentzi) crossing road. Identification and Descriptive Features: Tarantulas (Figures 1, 2, 3 and 6) are large, hairy spiders and all species found in Colorado are generally dark brown to black. Longer hairs are usually present on the abdomen and these may be a lighter brown color. Some banding of colors may be present on the legs. The carapace (back of the section with legs and head/cephalothorax) of eastern Colorado species ranges from light gray-brown to dark reddish brown with some coppery tones. Almost all tarantulas that are observed are mature males migrating in late summer and early fall. Females (Figure 2), which are much less commonly collected and observed than the adult males, will be found within the very near vicinity of their burrows. They usually have similar markings to the males but do not develop the very long legs of mature males. Adult females are also considerably larger than the Figure 2. Tarantula female emerging from males. For example, the carapace length of A. hentzi burrow. Otero County. may be about 20 mm in the female and only about 15 mm in the male. Two “mini-tarantulas” are known from western Colorado. Most common is Aphonopelma vogelae Smith (Figure 3). County records for this species include Montezuma, Montrose, and San Miguel. Males have a carapace length of about 10 mm. The western Colorado species tend to be more uniformly dark brown to black than those in southeastern Colorado. -

A Guide to Arthropods Bandelier National Monument
A Guide to Arthropods Bandelier National Monument Top left: Melanoplus akinus Top right: Vanessa cardui Bottom left: Elodes sp. Bottom right: Wolf Spider (Family Lycosidae) by David Lightfoot Compiled by Theresa Murphy Nov 2012 In collaboration with Collin Haffey, Craig Allen, David Lightfoot, Sandra Brantley and Kay Beeley WHAT ARE ARTHROPODS? And why are they important? What’s the difference between Arthropods and Insects? Most of this guide is comprised of insects. These are animals that have three body segments- head, thorax, and abdomen, three pairs of legs, and usually have wings, although there are several wingless forms of insects. Insects are of the Class Insecta and they make up the largest class of the phylum called Arthropoda (arthropods). However, the phylum Arthopoda includes other groups as well including Crustacea (crabs, lobsters, shrimps, barnacles, etc.), Myriapoda (millipedes, centipedes, etc.) and Arachnida (scorpions, king crabs, spiders, mites, ticks, etc.). Arthropods including insects and all other animals in this phylum are characterized as animals with a tough outer exoskeleton or body-shell and flexible jointed limbs that allow the animal to move. Although this guide is comprised mostly of insects, some members of the Myriapoda and Arachnida can also be found here. Remember they are all arthropods but only some of them are true ‘insects’. Entomologist - A scientist who focuses on the study of insects! What’s bugging entomologists? Although we tend to call all insects ‘bugs’ according to entomology a ‘true bug’ must be of the Order Hemiptera. So what exactly makes an insect a bug? Insects in the order Hemiptera have sucking, beak-like mouthparts, which are tucked under their “chin” when Metallic Green Bee (Agapostemon sp.) not in use. -

Husbandry Manual for Exotic Tarantulas
Husbandry Manual for Exotic Tarantulas Order: Araneae Family: Theraphosidae Author: Nathan Psaila Date: 13 October 2005 Sydney Institute of TAFE, Ultimo Course: Zookeeping Cert. III 5867 Lecturer: Graeme Phipps Table of Contents Introduction 6 1 Taxonomy 7 1.1 Nomenclature 7 1.2 Common Names 7 2 Natural History 9 2.1 Basic Anatomy 10 2.2 Mass & Basic Body Measurements 14 2.3 Sexual Dimorphism 15 2.4 Distribution & Habitat 16 2.5 Conservation Status 17 2.6 Diet in the Wild 17 2.7 Longevity 18 3 Housing Requirements 20 3.1 Exhibit/Holding Area Design 20 3.2 Enclosure Design 21 3.3 Spatial Requirements 22 3.4 Temperature Requirements 22 3.4.1 Temperature Problems 23 3.5 Humidity Requirements 24 3.5.1 Humidity Problems 27 3.6 Substrate 29 3.7 Enclosure Furnishings 30 3.8 Lighting 31 4 General Husbandry 32 4.1 Hygiene and Cleaning 32 4.1.1 Cleaning Procedures 33 2 4.2 Record Keeping 35 4.3 Methods of Identification 35 4.4 Routine Data Collection 36 5 Feeding Requirements 37 5.1 Captive Diet 37 5.2 Supplements 38 5.3 Presentation of Food 38 6 Handling and Transport 41 6.1 Timing of Capture and handling 41 6.2 Catching Equipment 41 6.3 Capture and Restraint Techniques 41 6.4 Weighing and Examination 44 6.5 Transport Requirements 44 6.5.1 Box Design 44 6.5.2 Furnishings 44 6.5.3 Water and Food 45 6.5.4 Release from Box 45 7 Health Requirements 46 7.1 Daily Health Checks 46 7.2 Detailed Physical Examination 47 7.3 Chemical Restraint 47 7.4 Routine Treatments 48 7.5 Known Health Problems 48 7.5.1 Dehydration 48 7.5.2 Punctures and Lesions 48 7.5.3 -

Araneae (Spider) Photos
Araneae (Spider) Photos Araneae (Spiders) About Information on: Spider Photos of Links to WWW Spiders Spiders of North America Relationships Spider Groups Spider Resources -- An Identification Manual About Spiders As in the other arachnid orders, appendage specialization is very important in the evolution of spiders. In spiders the five pairs of appendages of the prosoma (one of the two main body sections) that follow the chelicerae are the pedipalps followed by four pairs of walking legs. The pedipalps are modified to serve as mating organs by mature male spiders. These modifications are often very complicated and differences in their structure are important characteristics used by araneologists in the classification of spiders. Pedipalps in female spiders are structurally much simpler and are used for sensing, manipulating food and sometimes in locomotion. It is relatively easy to tell mature or nearly mature males from female spiders (at least in most groups) by looking at the pedipalps -- in females they look like functional but small legs while in males the ends tend to be enlarged, often greatly so. In young spiders these differences are not evident. There are also appendages on the opisthosoma (the rear body section, the one with no walking legs) the best known being the spinnerets. In the first spiders there were four pairs of spinnerets. Living spiders may have four e.g., (liphistiomorph spiders) or three pairs (e.g., mygalomorph and ecribellate araneomorphs) or three paris of spinnerets and a silk spinning plate called a cribellum (the earliest and many extant araneomorph spiders). Spinnerets' history as appendages is suggested in part by their being projections away from the opisthosoma and the fact that they may retain muscles for movement Much of the success of spiders traces directly to their extensive use of silk and poison. -

Arachnides 78
Arachnides n°78, 2016 ARACHNIDES BULLETIN DE TERRARIOPHILIE ET DE RECHERCHES DE L’A.P.C.I. (Association Pour la Connaissance des Invertébrés) 78 2016 0 Arachnides n°78, 2016 GRADIENTS DE LATITUDINALITÉ CHEZ LES SCORPIONS (ARACHNIDA: SCORPIONES). G. DUPRÉ Résumé. La répartition des communautés animales et végétales s'effectue selon un gradient latitudinal essentiellement climatique des pôles vers l'équateur. Les écosystèmes terrestres sont très variés et peuvent être des forêts boréales, des forêts tempérés, des forêts méditerranéennes, des déserts, des savanes ou encore des forêts tropicales. De nombreux auteurs admettent qu'un gradient de biodiversité s'accroit des pôles vers l'équateur (Sax, 2001; Boyero, 2006; Hallé, 2010). Nous avons tenté de vérifier ce fait pour les scorpions. Introduction. La répartition latitudinale des scorpions dans le monde se situe entre les latitudes 50° nord et 55° sud (Fig.1). Aucune étude n'a été entreprise pour préciser cette répartition à l'intérieur de ces limites. Ce vaste territoire englobe des biomes latitudinaux très différents y compris d'un continent à l'autre. Par exemple les zones désertiques en Australie ne sont pas situées aux mêmes latitudes que les zones désertiques africaines. A la même latitude on peut trouver la forêt tropicale amazonienne et la savane du Kénya, ce qui bien sûr implique une faune scorpionique écologiquement bien différente. Les résultats de cette étude sont présentés avec un certain nombre de difficultés discutées ci-après. Fig. 1. Carte de répartition mondiale des scorpions. Matériel et méthodes. L'étude a été arrêtée au 20 août 2016 donc sans tenir compte des espèces décrites après cette date. -

Spider Notebooking Pages Bw
Spider Notebooking Pages 2019 Stacey Jones at Simple Living. Creative Learning PINTEREST! | WEBSITE | FACEBOOK All rights reserved. No part of this book may be reproduced, stored or transmitted in any form by any means without prior permission of the publisher. This workbook is licensed for personal/family use only. YOU MAY: ! Use these files for personal use only. ! Use in your personal classroom ! Download the files to your personal computer. ! Print as many copies as you would like to use for your personal use. ! Direct other to our website: https://simplelivingcreativelearning.com YOU MAY NOT: ! Edit any of these printables. ! Share the files with anyone else. ! Store or sell them on any website. ! Claim them as your own. ! Print and sell or distribute them to others Graphics and Font: Spider Notebooking Pages Spider Notebooking Pages Spider Notebooking Pages By: !!!!!!!!!!!!!!!!!!!!!!!! Spider Notebooking Pages By: !!!!!!!!!!!!!!!!!!!!!!!! Spider Notebooking Pages By: !!!!!!!!!!!!!!!!!!!!!!!! Arrow-shaped micrathena (Micrathena sagittata) Diet Habitat Characteristics Simple Living. Creative Learning ! Arrow-shaped micrathena (Micrathena sagittata) ______________ ________0__000 00000000000000 Scientific Classification Order: ______________ ________________________ Suborder: ____________ Genus: _______________ ________________________ Species: _____________ ________________________ ________________________ ________________________ ________________________ ________________________ ________________________ ________________________ ________________________