G2 Phase Cell Cycle Regulation by E2F4 Following Genotoxic Stress
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Individual Index
INDIVIDUAL INDEX Bl Page Page A Cooremans-Cruz, Eugenio Juan 3825 Cortes, Eugenia 2474 Abbott, Charles P 3807 Crowley, Daniel 3820 Abrams, Samuel S 3825 Cruz, Marinelle Khristy 3836 Abshire, Arthur J 3812 Cuervo, Ester 3825 Ah Kwang Chang 3827 Cuffy, Jackson Ormiston Edwards and Ah Sing Ying 3826 Merle Cleopatra Edwards 38J54 Ah Young Cho Kwak 3803 Alcala-Salcedo, Apolinario 3825 D Alvarado, Irma Victoria Bolarte 3853 Araujo,Lilia 3828 D'Souza, Eustace John 3824 Araujo, Maria Freitas 3825 Datronics Engineers, Inc 3857 Arellano, Santos Marquez 3830 Daves, Gary 3862 Armstrong, Anne L 235 Davies, Mrs. Olive M.V.T., Samira Arriola, Rodolfo N 3851 D.K., Ola-Tomi K., Ola-Yinka K., Asencio-Placencio, Pedro 3825 Ilesha E.K., and Baba-Tunji K 3803 Atherley, Juana Todd 3829 Davis, Jefferson F 1304 Austin, John P 234 Day, Craig 3824 DeBravo, Cenovia Mesa 3825 B DeLuna-Segovia, Jose 3825 Dibben, James William 3823 Bader, Louis William 3825 Din, Ruben P 3831 Baglieri, George 3825 Do Sook Park 3841 Bakierowska, Kazimiera 3827 Dom Min Lee 3826 Bally, George Ally 3827 Dowd, Kazuko Nishioka 3820 Barengo, Bernardino 2485 Barrera-Cabrera, Jesus 3825 Batavia Turf Farms, Inc 3818 • ^- • E Benney, Donna Marainne 3836 Eaker, Lt. Gen. IraC 1060 Berger, Harry 3825 Eha,Elmar 3825 Binabise, Juanita 3840 Ehard,Karin 3858 Birely, Victor M 1643 Elder, William J 3814 Blakeley, Hildegard G 3845 Emde, Noel Abueg 3837 Blanquicett, Carmen Cecilia 3859 Encomienda, Rogelio M 3853 Bonderenko, Edward J 2485 Espenueva, Edmundo Alfredo Boone, Master Sgt. William E 3813 Oreiro -
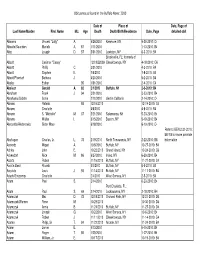
Obituaries Buffalo News 2010 by Name
Obituaries as found in the Buffalo News: 2010 Date of Place of Date, Page of Last Name/Maiden First Name M.I. Age Death Death/Birth/Residence Date, Page detailed obit Abbarno Vincent "Lolly" A. 9/26/2010 Kenmore, NY 9-30-2010: C4 Abbatte/Saunders Murielle A. 87 1/11/2010 1-13-2010: B4 Abbo Joseph D. 57 5/31/2010 Lewiston, NY 6-3-2010: B4 Brooksville, FL; formerly of Abbott Casimer "Casey" 12/19/22009 Cheektowaga, NY 4-18-2010: C6 Abbott Phillip C. 3/31/2010 4-3-2010: B4 Abbott Stephen E. 7/6/2010 7-8-2010: B4 Abbott/Pfoetsch Barbara J. 4/20/2010 5-2-2010: B4 Abeles Esther 95 1/31/2010 2-4-2010: C4 Abelson Gerald A. 82 2/1/2010 Buffalo, NY 2-3-2010: B4 Abraham Frank J. 94 3/21/2010 3-23-2010: B4 Abrahams/Gichtin Sonia 2/10/2010 died in California 2-14-2010: C4 Abramo Rafeala 93 12/16/2010 12-19-2010: C4 Abrams Charlotte 4/6/2010 4-8-2010: B4 Abrams S. "Michelle" M. 37 5/21/2010 Salamanca, NY 5-23-2010: B4 Abrams Walter I. 5/15/2010 Basom, NY 5-19-2010: B4 Abrosette/Aksterowicz Sister Mary 6/18/2010 6-19-2010: C4 Refer to BEN 2-21-2010: B6/7/8 for more possible Abshagen Charles, Jr. L. 73 2/19/2010 North Tonawanda, NY 2-22-2010: B8 information Acevedo Miguel A. 10/6/2010 Buffalo, NY 10-27-2010: B4 Achkar John E. -

Men Final Entries
Final Entries - Athletes List by event European Athletics Indoor Championships 2021, Torun/POL Tot. Number of countries Tot. Number of athletes Tot. Number of Men Tot. Number of Women 47 733 405 328 FINAL ENTRIES - Men 60m Senior Men Num. of countries: 33 Num. of athletes: 71 Member Federation Surname First Name DoB PB SB ARM Donigian Alexander 20/10/1993 6.64i 6.79i ART Keletela Dorian Celeste 06/02/1999 6.79i 6.85i AUT Fuchs Markus 14/11/1995 6.62i 6.69i BEL Kuba Di-Vita Gaylord 17/11/1995 6.73i 6.75i BEL Vleminckx Kobe 31/05/1998 6.65i 6.65i BLR Bliznets Dzianis 12/03/1995 6.75i 6.75i BLR Bohdan Maksim 19/03/1997 6.77i 6.77i BLR Zabalotny Yury 24/02/1997 6.72i 6.72i BUL Dimitrov Denis 10/02/1994 6.65i 6.73i BUL Jivkov Vesselin 26/01/2001 6.76i 6.80i CZE Hampl Štěpán 10/11/1999 6.70i 6.70i CZE Stromšík Zdeněk 25/11/1994 6.60i 6.68i CZE Veleba Jan 06/12/1986 6.65i 6.65i DEN Hansen Simon 30/06/1998 6.75i 6.75i DEN Kjær Emil Mader 20/12/1999 6.77i 6.77i DEN Musah Kojo 15/04/1996 6.61i 6.61i ESP López Sergio 05/07/1999 6.67i 6.74i ESP Rodríguez Daniel 26/01/1995 6.67i 6.67i ESP Sanchez Ricardo 10/08/1999 6.75i 6.75i EST Nazarov Karl Erik 17/03/1999 6.63i 6.63i FIN Illukka Riku 21/09/1999 6.73i 6.73i FIN Purola Samuel 19/05/2000 6.67i 6.67i FIN Samuelsson Samuli 23/06/1995 6.66i 6.66i FRA Fall Mouhamadou 25/02/1992 6.62i 6.62i FRA Golitin Amaury 28/01/1997 6.62i 6.62i GBR Aikines-Aryeetey Harry 29/08/1988 6.55i 6.67i GBR Bromby Oliver 30/03/1998 6.63i 6.65i GBR Robertson Andrew 17/12/1990 6.54i 6.61i GER Corucle Philipp 18/07/1997 6.62i -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Supplementary Data
SUPPLEMENTARY DATA A cyclin D1-dependent transcriptional program predicts clinical outcome in mantle cell lymphoma Santiago Demajo et al. 1 SUPPLEMENTARY DATA INDEX Supplementary Methods p. 3 Supplementary References p. 8 Supplementary Tables (S1 to S5) p. 9 Supplementary Figures (S1 to S15) p. 17 2 SUPPLEMENTARY METHODS Western blot, immunoprecipitation, and qRT-PCR Western blot (WB) analysis was performed as previously described (1), using cyclin D1 (Santa Cruz Biotechnology, sc-753, RRID:AB_2070433) and tubulin (Sigma-Aldrich, T5168, RRID:AB_477579) antibodies. Co-immunoprecipitation assays were performed as described before (2), using cyclin D1 antibody (Santa Cruz Biotechnology, sc-8396, RRID:AB_627344) or control IgG (Santa Cruz Biotechnology, sc-2025, RRID:AB_737182) followed by protein G- magnetic beads (Invitrogen) incubation and elution with Glycine 100mM pH=2.5. Co-IP experiments were performed within five weeks after cell thawing. Cyclin D1 (Santa Cruz Biotechnology, sc-753), E2F4 (Bethyl, A302-134A, RRID:AB_1720353), FOXM1 (Santa Cruz Biotechnology, sc-502, RRID:AB_631523), and CBP (Santa Cruz Biotechnology, sc-7300, RRID:AB_626817) antibodies were used for WB detection. In figure 1A and supplementary figure S2A, the same blot was probed with cyclin D1 and tubulin antibodies by cutting the membrane. In figure 2H, cyclin D1 and CBP blots correspond to the same membrane while E2F4 and FOXM1 blots correspond to an independent membrane. Image acquisition was performed with ImageQuant LAS 4000 mini (GE Healthcare). Image processing and quantification were performed with Multi Gauge software (Fujifilm). For qRT-PCR analysis, cDNA was generated from 1 µg RNA with qScript cDNA Synthesis kit (Quantabio). qRT–PCR reaction was performed using SYBR green (Roche). -

Proteogenomic Analysis of Inhibitor of Differentiation 4 (ID4) in Basal-Like Breast Cancer Laura A
Baker et al. Breast Cancer Research (2020) 22:63 https://doi.org/10.1186/s13058-020-01306-6 RESEARCH ARTICLE Open Access Proteogenomic analysis of Inhibitor of Differentiation 4 (ID4) in basal-like breast cancer Laura A. Baker1,2, Holly Holliday1,2†, Daniel Roden1,2†, Christoph Krisp3,4†, Sunny Z. Wu1,2, Simon Junankar1,2, Aurelien A. Serandour5, Hisham Mohammed5, Radhika Nair6, Geetha Sankaranarayanan5, Andrew M. K. Law1,2, Andrea McFarland1, Peter T. Simpson7, Sunil Lakhani7,8, Eoin Dodson1,2, Christina Selinger9, Lyndal Anderson9,10, Goli Samimi11, Neville F. Hacker12, Elgene Lim1,2, Christopher J. Ormandy1,2, Matthew J. Naylor13, Kaylene Simpson14,15, Iva Nikolic14, Sandra O’Toole1,2,9,10, Warren Kaplan1, Mark J. Cowley1,2, Jason S. Carroll5, Mark Molloy3 and Alexander Swarbrick1,2* Abstract Background: Basal-like breast cancer (BLBC) is a poorly characterised, heterogeneous disease. Patients are diagnosed with aggressive, high-grade tumours and often relapse with chemotherapy resistance. Detailed understanding of the molecular underpinnings of this disease is essential to the development of personalised therapeutic strategies. Inhibitor of differentiation 4 (ID4) is a helix-loop-helix transcriptional regulator required for mammary gland development. ID4 is overexpressed in a subset of BLBC patients, associating with a stem-like poor prognosis phenotype, and is necessary for the growth of cell line models of BLBC through unknown mechanisms. Methods: Here, we have defined unique molecular insights into the function of ID4 in BLBC and the related disease high-grade serous ovarian cancer (HGSOC), by combining RIME proteomic analysis, ChIP-seq mapping of genomic binding sites and RNA-seq. -

Cytoplasmic E2f4 Forms Organizing Centres for Initiation of Centriole Amplification During Multiciliogenesis
ARTICLE Received 13 Feb 2017 | Accepted 8 May 2017 | Published 4 Jul 2017 DOI: 10.1038/ncomms15857 OPEN Cytoplasmic E2f4 forms organizing centres for initiation of centriole amplification during multiciliogenesis Munemasa Mori1, Renin Hazan2, Paul S. Danielian2, John E. Mahoney1,w, Huijun Li1, Jining Lu1, Emily S. Miller2, Xueliang Zhu3, Jacqueline A. Lees2 & Wellington V. Cardoso1 Abnormal development of multiciliated cells is a hallmark of a variety of human conditions associated with chronic airway diseases, hydrocephalus and infertility. Multiciliogenesis requires both activation of a specialized transcriptional program and assembly of cytoplasmic structures for large-scale centriole amplification that generates basal bodies. It remains unclear, however, what mechanism initiates formation of these multiprotein complexes in epithelial progenitors. Here we show that this is triggered by nucleocytoplasmic translocation of the transcription factor E2f4. After inducing a transcriptional program of centriole biogenesis, E2f4 forms apical cytoplasmic organizing centres for assembly and nucleation of deuterosomes. Using genetically altered mice and E2F4 mutant proteins we demonstrate that centriole amplification is crucially dependent on these organizing centres and that, without cytoplasmic E2f4, deuterosomes are not assembled, halting multiciliogenesis. Thus, E2f4 integrates nuclear and previously unsuspected cytoplasmic events of centriole amplification, providing new perspectives for the understanding of normal ciliogenesis, ciliopathies and cancer. 1 Columbia Center for Human Development, Department of Medicine, Pulmonary Allergy Critical Care, Columbia University Medical Center, New York City, New York 10032, USA. 2 David H. Koch Institute for Integrative Cancer Research, MIT, Cambridge, Massachusetts 02139, USA. 3 State Key Laboratory of Cell Biology, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yueyang Road, Shanghai 200031, China. -

Sales Report
Sales Listing Report Page 1 of 262 McHenry County 01/21/2021 08:25:40 01/01/2020 - 09/30/2020 Township: GRAFTON TWP Document Number Sale Year Sale Type Valid Sale Sale Date Dept. Study Selling Price Parcel Number Built Year Property Type Prop. Class Acres Square Ft. Lot Size Grantor Name Grantee Name Site Address 2020R0006895 2020 Warranty Deed Y 02/13/2020 Y $189,000.00 REINVEST HOMES, LLC ANGELA STROPE 160 S HEATHER DR 18-01-101-027 0 GARAGE/ NO O O 0040 .00 0 CRYSTAL LAKE, IL 600145177 160 S HEATHER DR CRYSTAL LAKE, IL 60014 - Legal Description: DOC 2020R0006895 LT 31 BLK 15 R A CEPEKS CRYSTAL VISTA 2020R0030830 2020 Partial Assessmen N 07/30/2020 N $136,500.00 DANIEL M ADAMS, AS TRUST BRETT A RIDINGS 1324 THORNWOOD LN 18-01-102-037 0 GARAGE/ NO O O 0040 .00 0 CRYSTAL LAKE, IL 600145042 TARI D RIDINGS 1324 THORNWOOD LANE CRYSTAL LAKE, IL 600140000 1324 THORNWOOD LN CRYSTAL LAKE, IL 60014 -5042 Legal Description: DOC 2020R0030830 LT 1 & PT LT 19 LYING N OF & ADJ BLK 14 R A CEPEKS CRYSTAL VISTA 2020R0010709 2020 Warranty Deed Y 03/16/2020 Y $210,000.00 PATRICK J. CARAMELA JUAN P. MIRANDA STACY C. CARAMELA 58 HOLLY DR 18-01-106-018 0 LDG SINGLE FAM 0040 .00 0 CRYSTAL LAKE, IL 600145022 58 HOLLY DR CRYSTAL LAKE, IL 60014 -5022 Legal Description: DOC 2020R0010709 LT 7 BLK 19 R A CEPEKS CRYSTAL VISTA 2020R0015717 2020 Warranty Deed Y 04/23/2020 Y $95,000.00 LISA M SULMA FANO THEOFANOUS 5011 NORTHWEST HWY 18-01-107-011 0 LDG SINGLE FAM 0040 .00 0 CRYSTAL LAKE, IL 600147330 STEVE THEOFANOUS 5011 NORTHWEST HIGHWAY CRYSTAL LAKE, IL 600147330 113 HEATHER DR CRYSTAL LAKE, IL 60014 - Legal Description: DOC 2020R0015717 LT 12 BLK 31 R A CEPEKS CRYSTAL VISTA Copyright (C) 1997-2021 DEVNET Incorporated MXSTIEG Sales Listing Report Page 2 of 262 McHenry County 01/21/2021 08:25:40 01/01/2020 - 09/30/2020 Township: GRAFTON TWP Document Number Sale Year Sale Type Valid Sale Sale Date Dept. -
![Arxiv:1810.00224V2 [Q-Bio.PE] 7 Dec 2020 Humanity Is Increasingly Influencing Global Environments [195]](https://docslib.b-cdn.net/cover/3556/arxiv-1810-00224v2-q-bio-pe-7-dec-2020-humanity-is-increasingly-in-uencing-global-environments-195-943556.webp)
Arxiv:1810.00224V2 [Q-Bio.PE] 7 Dec 2020 Humanity Is Increasingly Influencing Global Environments [195]
A Survey of Biodiversity Informatics: Concepts, Practices, and Challenges Luiz M. R. Gadelha Jr.1* Pedro C. de Siracusa1 Artur Ziviani1 Eduardo Couto Dalcin2 Helen Michelle Affe2 Marinez Ferreira de Siqueira2 Luís Alexandre Estevão da Silva2 Douglas A. Augusto3 Eduardo Krempser3 Marcia Chame3 Raquel Lopes Costa4 Pedro Milet Meirelles5 and Fabiano Thompson6 1National Laboratory for Scientific Computing, Petrópolis, Brazil 2Friedrich-Schiller-University Jena, Jena, Germany 2Rio de Janeiro Botanical Garden, Rio de Janeiro, Brazil 3Oswaldo Cruz Foundation, Rio de Janeiro, Brazil 4National Institute of Cancer, Rio de Janeiro, Brazil 5Federal University of Bahia, Salvador, Brazil 6Federal University of Rio de Janeiro, Rio de Janeiro, Brazil Abstract The unprecedented size of the human population, along with its associated economic activities, have an ever increasing impact on global environments. Across the world, countries are concerned about the growing resource consumption and the capacity of ecosystems to provide them. To effectively conserve biodiversity, it is essential to make indicators and knowledge openly available to decision-makers in ways that they can effectively use them. The development and deployment of mechanisms to produce these indicators depend on having access to trustworthy data from field surveys and automated sensors, biological collections, molec- ular data, and historic academic literature. The transformation of this raw data into synthesized information that is fit for use requires going through many refinement steps. The methodologies and techniques used to manage and analyze this data comprise an area often called biodiversity informatics (or e-Biodiversity). Bio- diversity data follows a life cycle consisting of planning, collection, certification, description, preservation, discovery, integration, and analysis. -

Table of Contents Methods
SUPPLEMENTARY MATERIAL FOR: Intrinsic specificity differences between transcription factor paralogs partly explain their differential in vivo binding Ning Shen1,2,3, Jingkang Zhao1,3,4, Joshua Schipper1,3, Yuning Zhang1,4,Tristan Bepler1, Dan Leehr1, John Bradley1, John Horton1,3, Hilmar Lapp1, Raluca Gordan1,3,5* 1Center for Genomic and Computational Biology, 2Department of Pharmacology and Cancer Biology, 3Department of Biostatistics and Bioinformatics, 4Program in Computational Biology and Bioinformatics, 5Department of Computer Science, Duke University, Durham, NC 27708 * Corresponding author. E-mail: [email protected] Table of Contents Methods ................................................................................................................................................... 1 1. ChIP-seq and DNase-seq data ................................................................................................................. 1 2. Design of DNA libraries for gcPBM assays .......................................................................................... 1 3. Protein expression and purification .................................................................................................... 3 4. Universal and genomic-context PBM assays ..................................................................................... 4 5. gcPBM data processing. Identifying core motifs .............................................................................. 4 6. Building core-stratified SVR models of TF-DNA binding specificity -
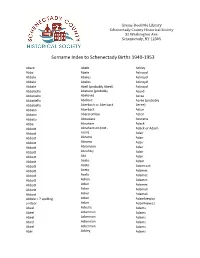
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave.