The Critical Role of Histone Methylation in Tumour Progression and As Anti-Cancer Targets in Neuroblastoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Cross-Talk Between Methylation and Phosphorylation in Lymphoid-Specific Helicase Drives Cancer Stem-Like Properties
Signal Transduction and Targeted Therapy www.nature.com/sigtrans ARTICLE OPEN The cross-talk between methylation and phosphorylation in lymphoid-specific helicase drives cancer stem-like properties Na Liu1,2,3, Rui Yang1,2, Ying Shi1,2, Ling Chen1,2, Yating Liu1,2, Zuli Wang1,2, Shouping Liu1,2, Lianlian Ouyang4, Haiyan Wang1,2, Weiwei Lai1,2, Chao Mao1,2, Min Wang1,2, Yan Cheng5, Shuang Liu4, Xiang Wang6, Hu Zhou7, Ya Cao1,2, Desheng Xiao1 and Yongguang Tao1,2,6 Posttranslational modifications (PTMs) of proteins, including chromatin modifiers, play crucial roles in the dynamic alteration of various protein properties and functions including stem-cell properties. However, the roles of Lymphoid-specific helicase (LSH), a DNA methylation modifier, in modulating stem-like properties in cancer are still not clearly clarified. Therefore, exploring PTMs modulation of LSH activity will be of great significance to further understand the function and activity of LSH. Here, we demonstrate that LSH is capable to undergo PTMs, including methylation and phosphorylation. The arginine methyltransferase PRMT5 can methylate LSH at R309 residue, meanwhile, LSH could as well be phosphorylated by MAPK1 kinase at S503 residue. We further show that the accumulation of phosphorylation of LSH at S503 site exhibits downregulation of LSH methylation at R309 residue, which eventually promoting stem-like properties in lung cancer. Whereas, phosphorylation-deficient LSH S503A mutant promotes the accumulation of LSH methylation at R309 residue and attenuates stem-like properties, indicating the critical roles of LSH PTMs in modulating stem-like properties. Thus, our study highlights the importance of the crosstalk between LSH PTMs in determining its activity and function in lung cancer stem-cell maintenance. -

Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression
International Journal of Molecular Sciences Review Hypoxia and Oxygen-Sensing Signaling in Gene Regulation and Cancer Progression Guang Yang, Rachel Shi and Qing Zhang * Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; [email protected] (G.Y.); [email protected] (R.S.) * Correspondence: [email protected]; Tel.: +1-214-645-4671 Received: 6 October 2020; Accepted: 29 October 2020; Published: 31 October 2020 Abstract: Oxygen homeostasis regulation is the most fundamental cellular process for adjusting physiological oxygen variations, and its irregularity leads to various human diseases, including cancer. Hypoxia is closely associated with cancer development, and hypoxia/oxygen-sensing signaling plays critical roles in the modulation of cancer progression. The key molecules of the hypoxia/oxygen-sensing signaling include the transcriptional regulator hypoxia-inducible factor (HIF) which widely controls oxygen responsive genes, the central members of the 2-oxoglutarate (2-OG)-dependent dioxygenases, such as prolyl hydroxylase (PHD or EglN), and an E3 ubiquitin ligase component for HIF degeneration called von Hippel–Lindau (encoding protein pVHL). In this review, we summarize the current knowledge about the canonical hypoxia signaling, HIF transcription factors, and pVHL. In addition, the role of 2-OG-dependent enzymes, such as DNA/RNA-modifying enzymes, JmjC domain-containing enzymes, and prolyl hydroxylases, in gene regulation of cancer progression, is specifically reviewed. We also discuss the therapeutic advancement of targeting hypoxia and oxygen sensing pathways in cancer. Keywords: hypoxia; PHDs; TETs; JmjCs; HIFs 1. Introduction Molecular oxygen serves as a co-factor in many biochemical processes and is fundamental for aerobic organisms to maintain intracellular ATP levels [1,2]. -

Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy
Cancers 2011, 3, 1691-1707; doi:10.3390/cancers3021691 OPEN ACCESS cancers ISSN 2072-6694 www.mdpi.com/journal/cancers Review Epigenetics of Estrogen Receptor Signaling: Role in Hormonal Cancer Progression and Therapy Monica Mann 1, Valerie Cortez 1 and Ratna K. Vadlamudi 2,* 1 Department of Cellular and Structural Biology, UTHSCSA, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA; E-Mails: [email protected] (M.M.); [email protected] (V.C.) 2 Department of Obstetrics and Gynecology, UTHSCSA, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-210-567-4930; Fax: +1-210-567-4958. Received: 4 January 2011; in revised form: 11 March 2011 / Accepted: 25 March 2011 / Published: 29 March 2011 Abstract: Estrogen receptor (ER) signaling plays a key role in hormonal cancer progression. ER is a ligand-dependent transcription factor that modulates gene transcription via recruitment to the target gene chromatin. Emerging evidence suggests that ER signaling has the potential to contribute to epigenetic changes. Estrogen stimulation is shown to induce several histone modifications at the ER target gene promoters including acetylation, phosphorylation and methylation via dynamic interactions with histone modifying enzymes. Deregulation of enzymes involved in the ER-mediated epigenetic pathway could play a vital role in ER driven neoplastic processes. Unlike genetic alterations, epigenetic changes are reversible, and hence offer novel therapeutic opportunities to reverse ERdriven epigenetic changes In this review, we summarize current knowledge on mechanisms by which ER signaling potentiates epigenetic changes in cancer cells via histone modifications. -

G2 Phase Cell Cycle Regulation by E2F4 Following Genotoxic Stress
G2 Phase Cell Cycle Regulation by E2F4 Following Genotoxic Stress by MEREDITH ELLEN CROSBY Submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Thesis Advisor: Dr. Alex Almasan Department of Environmental Health Sciences CASE WESTERN RESERVE UNIVERSITY May, 2006 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the dissertation of ______________________________________________________ candidate for the Ph.D. degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. TABLE OF CONTENTS TABLE OF CONTENTS………………………………………………………………….1 LIST OF FIGURES……………………………………………………………………….5 LIST OF TABLES………………………………………………………………………...7 ACKNOWLEDGEMENTS……………………………………………………………….8 LIST OF ABBREVIATIONS……………………………………………………………10 ABSTRACT……………………………………………………………………………...15 CHAPTER 1. INTRODUCTION 1.1. CELL CYCLE REGULATION: HISTORICAL OVERVIEW………………...17 1.2. THE E2F FAMILY OF TRANSCRIPTION FACTORS……………………….22 1.3. E2F AND CELL CYCLE CONTROL 1.3.1. G0/G1 Phase Transition………………………………………………….28 1.3.2. S Phase…………………………………………………………………...28 1.3.3. G2/M Phase Transition…………………………………………………..30 1.4. -

Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2010 Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis Ryan Fassnacht Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Physiology Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/2246 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Ryan C. Fassnacht 2010 All Rights Reserved Molecular Mechanisms Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University. by Ryan Christopher Fassnacht, B.S. Hampden Sydney University, 2005 M.S. Virginia Commonwealth University, 2010 Director: Valeria Mas, Ph.D., Associate Professor of Surgery and Pathology Division of Transplant Department of Surgery Virginia Commonwealth University Richmond, Virginia July 9, 2010 Acknowledgement The Author wishes to thank his family and close friends for their support. He would also like to thank the members of the molecular transplant team for their help and advice. This project would not have been possible with out the help of Dr. Valeria Mas and her endearing -

Individual Index
INDIVIDUAL INDEX Bl Page Page A Cooremans-Cruz, Eugenio Juan 3825 Cortes, Eugenia 2474 Abbott, Charles P 3807 Crowley, Daniel 3820 Abrams, Samuel S 3825 Cruz, Marinelle Khristy 3836 Abshire, Arthur J 3812 Cuervo, Ester 3825 Ah Kwang Chang 3827 Cuffy, Jackson Ormiston Edwards and Ah Sing Ying 3826 Merle Cleopatra Edwards 38J54 Ah Young Cho Kwak 3803 Alcala-Salcedo, Apolinario 3825 D Alvarado, Irma Victoria Bolarte 3853 Araujo,Lilia 3828 D'Souza, Eustace John 3824 Araujo, Maria Freitas 3825 Datronics Engineers, Inc 3857 Arellano, Santos Marquez 3830 Daves, Gary 3862 Armstrong, Anne L 235 Davies, Mrs. Olive M.V.T., Samira Arriola, Rodolfo N 3851 D.K., Ola-Tomi K., Ola-Yinka K., Asencio-Placencio, Pedro 3825 Ilesha E.K., and Baba-Tunji K 3803 Atherley, Juana Todd 3829 Davis, Jefferson F 1304 Austin, John P 234 Day, Craig 3824 DeBravo, Cenovia Mesa 3825 B DeLuna-Segovia, Jose 3825 Dibben, James William 3823 Bader, Louis William 3825 Din, Ruben P 3831 Baglieri, George 3825 Do Sook Park 3841 Bakierowska, Kazimiera 3827 Dom Min Lee 3826 Bally, George Ally 3827 Dowd, Kazuko Nishioka 3820 Barengo, Bernardino 2485 Barrera-Cabrera, Jesus 3825 Batavia Turf Farms, Inc 3818 • ^- • E Benney, Donna Marainne 3836 Eaker, Lt. Gen. IraC 1060 Berger, Harry 3825 Eha,Elmar 3825 Binabise, Juanita 3840 Ehard,Karin 3858 Birely, Victor M 1643 Elder, William J 3814 Blakeley, Hildegard G 3845 Emde, Noel Abueg 3837 Blanquicett, Carmen Cecilia 3859 Encomienda, Rogelio M 3853 Bonderenko, Edward J 2485 Espenueva, Edmundo Alfredo Boone, Master Sgt. William E 3813 Oreiro -

Histone Demethylase JMJD6 Regulates Cellular Migration And
Shen et al. Stem Cell Research & Therapy (2018) 9:212 https://doi.org/10.1186/s13287-018-0949-3 RESEARCH Open Access Histone demethylase JMJD6 regulates cellular migration and proliferation in adipose-derived mesenchymal stem cells Chongyang Shen1,2, Qingli Quan3*, Chuan Yang1, Yueqiang Wen2 and Hong Li1* Abstract Background: Adipose-derived mesenchymal stem cells (ADSCs) have been extensively explored as a promising therapeutic agent due to their differentiation, proliferation and migration abilities. The epigenetic mechanisms that regulate the fate of mesenchymal stem cells (MSCs) have been described in detail. However, the epigenetic modulation of ADSCs proliferation and migration is poorly understood. Methods: The present study examined histone demethylases roles and expression by RT-PCR, as well as through siRNA screening and ChIP-qPCR assay. Cellular proliferation and migration assays were employed in shRNA- mediated JMJD6 knockdown and control ADSCs. PDE1C inhibition studies were conducted to confirm its role in JMJD6-mediated epigenetic regulation of ADSCs. Results: The data demonstrate that the histone demethylase JMJD6 plays a critical role in regulating the proliferation and migration of ADSCs by removing H4R3me2a at the promoter regions of PDEC1 and suppressing PDEC1 expression. Importantly, the depletion of JMJD6 in ADSCs significantly increased cellular proliferation and motility, which was associated with increases in PDE1C expression and decreases in the levels of both cAMP and cGMP. The increase in proliferation and migration was reversed by treatment with a PDE1C inhibitor, suggesting that JMJD6 attenuates the proliferation and migration of ADSCs as an epigenetic regulator and PDE1C partially contributes to the JMJD6-mediated regulation. Conclusions: Taken together, our results indicate for the first time that JMJD6 plays an important role in the regulation of ADSCs proliferation and migration through the modulation of PDE1C expression. -

Preclinical Evaluation of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma by Andrea Shergalis
Preclinical Evaluation of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma By Andrea Shergalis A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Medicinal Chemistry) in the University of Michigan 2020 Doctoral Committee: Professor Nouri Neamati, Chair Professor George A. Garcia Professor Peter J. H. Scott Professor Shaomeng Wang Andrea G. Shergalis [email protected] ORCID 0000-0002-1155-1583 © Andrea Shergalis 2020 All Rights Reserved ACKNOWLEDGEMENTS So many people have been involved in bringing this project to life and making this dissertation possible. First, I want to thank my advisor, Prof. Nouri Neamati, for his guidance, encouragement, and patience. Prof. Neamati instilled an enthusiasm in me for science and drug discovery, while allowing me the space to independently explore complex biochemical problems, and I am grateful for his kind and patient mentorship. I also thank my committee members, Profs. George Garcia, Peter Scott, and Shaomeng Wang, for their patience, guidance, and support throughout my graduate career. I am thankful to them for taking time to meet with me and have thoughtful conversations about medicinal chemistry and science in general. From the Neamati lab, I would like to thank so many. First and foremost, I have to thank Shuzo Tamara for being an incredible, kind, and patient teacher and mentor. Shuzo is one of the hardest workers I know. In addition to a strong work ethic, he taught me pretty much everything I know and laid the foundation for the article published as Chapter 3 of this dissertation. The work published in this dissertation really began with the initial identification of PDI as a target by Shili Xu, and I am grateful for his advice and guidance (from afar!). -

The Dynamic Role of Jumonji C Domain Containing Protein 6 in Placental Development and Disease
The Dynamic Role of Jumonji C Domain Containing Protein 6 in Placental Development and Disease by Sruthi Alahari A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Physiology University of Toronto © Copyright by Sruthi Alahari 2017 i The Dynamic Role of Jumonji C Domain Containing Protein 6 in Placental Development and Disease Sruthi Alahari Doctor of Philosophy Department of Physiology University of Toronto 2017 Abstract Perturbations in oxygen sensing are a defining feature of placental-associated pathologies such as preeclampsia, a serious disorder of pregnancy. Preeclamptic placentae have markedly elevated levels of Hypoxia Inducible Factor 1α (HIF1A), a master regulator of oxygen homeostasis. Mounting evidence implicates a family of Fe2+ and oxygen-dependent Jumonji C domain containing enzymes (JMJDs) as mediators of the epigenetic code and hypoxic gene expression. While several JMJDs are induced in hypoxia, their role in pregnancy remains unclear. The goal of this study was to characterize JMJD6 function in the placenta in physiological and pathological conditions, and unravel its regulatory relationship with von Hippel Lindau tumour suppressor (VHL), a key executor of the cellular hypoxic response. JMJD6 expression inversely correlated with changes in oxygen tension during placental development, while JMJD6 protein and mRNA were significantly elevated in low oxygen and in early-onset preeclamptic (E-PE) placentae. In vitro demethylation assays revealed that optimal JMJD6-dependent demethylation of its histone targets, H3R2me2s and H4R3me2s, occurred in normoxia, and this was impaired in E-PE placentae due to a hypoxia-iron imbalance. In cytotrophoblast cells, JMJD6 is a positive ii regulator of VHL gene expression in normoxia. -
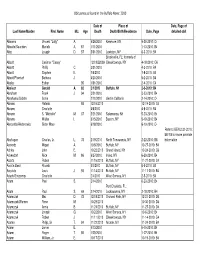
Obituaries Buffalo News 2010 by Name
Obituaries as found in the Buffalo News: 2010 Date of Place of Date, Page of Last Name/Maiden First Name M.I. Age Death Death/Birth/Residence Date, Page detailed obit Abbarno Vincent "Lolly" A. 9/26/2010 Kenmore, NY 9-30-2010: C4 Abbatte/Saunders Murielle A. 87 1/11/2010 1-13-2010: B4 Abbo Joseph D. 57 5/31/2010 Lewiston, NY 6-3-2010: B4 Brooksville, FL; formerly of Abbott Casimer "Casey" 12/19/22009 Cheektowaga, NY 4-18-2010: C6 Abbott Phillip C. 3/31/2010 4-3-2010: B4 Abbott Stephen E. 7/6/2010 7-8-2010: B4 Abbott/Pfoetsch Barbara J. 4/20/2010 5-2-2010: B4 Abeles Esther 95 1/31/2010 2-4-2010: C4 Abelson Gerald A. 82 2/1/2010 Buffalo, NY 2-3-2010: B4 Abraham Frank J. 94 3/21/2010 3-23-2010: B4 Abrahams/Gichtin Sonia 2/10/2010 died in California 2-14-2010: C4 Abramo Rafeala 93 12/16/2010 12-19-2010: C4 Abrams Charlotte 4/6/2010 4-8-2010: B4 Abrams S. "Michelle" M. 37 5/21/2010 Salamanca, NY 5-23-2010: B4 Abrams Walter I. 5/15/2010 Basom, NY 5-19-2010: B4 Abrosette/Aksterowicz Sister Mary 6/18/2010 6-19-2010: C4 Refer to BEN 2-21-2010: B6/7/8 for more possible Abshagen Charles, Jr. L. 73 2/19/2010 North Tonawanda, NY 2-22-2010: B8 information Acevedo Miguel A. 10/6/2010 Buffalo, NY 10-27-2010: B4 Achkar John E. -

Men Final Entries
Final Entries - Athletes List by event European Athletics Indoor Championships 2021, Torun/POL Tot. Number of countries Tot. Number of athletes Tot. Number of Men Tot. Number of Women 47 733 405 328 FINAL ENTRIES - Men 60m Senior Men Num. of countries: 33 Num. of athletes: 71 Member Federation Surname First Name DoB PB SB ARM Donigian Alexander 20/10/1993 6.64i 6.79i ART Keletela Dorian Celeste 06/02/1999 6.79i 6.85i AUT Fuchs Markus 14/11/1995 6.62i 6.69i BEL Kuba Di-Vita Gaylord 17/11/1995 6.73i 6.75i BEL Vleminckx Kobe 31/05/1998 6.65i 6.65i BLR Bliznets Dzianis 12/03/1995 6.75i 6.75i BLR Bohdan Maksim 19/03/1997 6.77i 6.77i BLR Zabalotny Yury 24/02/1997 6.72i 6.72i BUL Dimitrov Denis 10/02/1994 6.65i 6.73i BUL Jivkov Vesselin 26/01/2001 6.76i 6.80i CZE Hampl Štěpán 10/11/1999 6.70i 6.70i CZE Stromšík Zdeněk 25/11/1994 6.60i 6.68i CZE Veleba Jan 06/12/1986 6.65i 6.65i DEN Hansen Simon 30/06/1998 6.75i 6.75i DEN Kjær Emil Mader 20/12/1999 6.77i 6.77i DEN Musah Kojo 15/04/1996 6.61i 6.61i ESP López Sergio 05/07/1999 6.67i 6.74i ESP Rodríguez Daniel 26/01/1995 6.67i 6.67i ESP Sanchez Ricardo 10/08/1999 6.75i 6.75i EST Nazarov Karl Erik 17/03/1999 6.63i 6.63i FIN Illukka Riku 21/09/1999 6.73i 6.73i FIN Purola Samuel 19/05/2000 6.67i 6.67i FIN Samuelsson Samuli 23/06/1995 6.66i 6.66i FRA Fall Mouhamadou 25/02/1992 6.62i 6.62i FRA Golitin Amaury 28/01/1997 6.62i 6.62i GBR Aikines-Aryeetey Harry 29/08/1988 6.55i 6.67i GBR Bromby Oliver 30/03/1998 6.63i 6.65i GBR Robertson Andrew 17/12/1990 6.54i 6.61i GER Corucle Philipp 18/07/1997 6.62i -

(12) Patent Application Publication (10) Pub. No.: US 2015/0315643 A1 O'garra Et Al
US 201503 15643A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0315643 A1 O'Garra et al. (43) Pub. Date: Nov. 5, 2015 (54) BLOODTRANSCRIPTIONAL SIGNATURES Publication Classification OF ACTIVE PULMONARY TUBERCULOSS AND SARCODOSIS (51) Int. C. CI2O I/68 (2006.01) (71) Applicants: BAYLOR RESEARCH INSTITUTE, (52) U.S. C. Dallas, TX (US); MEDICAL CPC ............ CI2O I/6883 (2013.01): CI2O I/6886 RESEARCH COUNCIL, Swindon (2013.01); C12O 2600/158 (2013.01); C12O (GB); IMPERIAL COLLEGE 2600/16 (2013.01); C12O 2600/1 18 (2013.01); HEALTHCARE NHS TRUST, London C12O 2600/1 12 (2013.01); C12O 2600/106 (GB) (2013.01) (72) Inventors: Anne O'Garra, London (GB); Chloe (57) ABSTRACT Bloom, London (GB); Matthew Paul The present invention includes a method of determining a Reddoch Berry, London (GB); Jacques lung disease from a patient Suspected of sarcoidosis, tuber F. Banchereau, Montclair, NJ (US); culosis, lung cancer or pneumonia comprising: obtaining a Damien Chaussabel, Bainbridge Island, sample from whole blood of the patient suspected of sarcoi WA (US); Viginia Maria Pascual, dosis, tuberculosis, lung cancer or pneumonia; detecting Dallas, TX (US) expression of (although not exclusive) six or more disease (21) Appl. No.: 14/651,989 genes, markers, or probes selected from SEQID NOS.: 1 to 1446, wherein increased expression of mRNA of upregulated (22) PCT Fled: Dec. 13, 2013 sarcoidosis, tuberculosis, lung cancer and pneumonia mark ers of SEQID NOS.: 1 to 1446 and/or decreased expression (86) PCT NO.: PCT/US2013/075097 of mRNA of downregulated Sarcoidosis, tuberculosis, lung cancer or pneumonia markers of SEQ ID NOS.: 1 to 1446 S371 (c)(1), relative to the expression of the mRNAs from a normal (2) Date: Jun.