Downloaded from the NCBI FTP Site [37]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -

Review Article: Inhibition of Methanogenic Archaea by Statins As a Targeted Management Strategy for Constipation and Related Disorders
Alimentary Pharmacology and Therapeutics Review article: inhibition of methanogenic archaea by statins as a targeted management strategy for constipation and related disorders K. Gottlieb*, V. Wacher*, J. Sliman* & M. Pimentel† *Synthetic Biologics, Inc., Rockville, SUMMARY MD, USA. † Gastroenterology, Cedars-Sinai Background Medical Center, Los Angeles, CA, USA. Observational studies show a strong association between delayed intestinal transit and the production of methane. Experimental data suggest a direct inhibitory activity of methane on the colonic and ileal smooth muscle and Correspondence to: a possible role for methane as a gasotransmitter. Archaea are the only con- Dr K. Gottlieb, Synthetic Biologics, fi Inc., 9605 Medical Center Drive, rmed biological sources of methane in nature and Methanobrevibacter Rockville, MD 20850, USA. smithii is the predominant methanogen in the human intestine. E-mail: [email protected] Aim To review the biosynthesis and composition of archaeal cell membranes, Publication data archaeal methanogenesis and the mechanism of action of statins in this context. Submitted 8 September 2015 First decision 29 September 2015 Methods Resubmitted 7 October 2015 Narrative review of the literature. Resubmitted 20 October 2015 Accepted 20 October 2015 Results EV Pub Online 11 November 2015 Statins can inhibit archaeal cell membrane biosynthesis without affecting This uncommissioned review article was bacterial numbers as demonstrated in livestock and humans. This opens subject to full peer-review. the possibility of a therapeutic intervention that targets a specific aetiologi- cal factor of constipation while protecting the intestinal microbiome. While it is generally believed that statins inhibit methane production via their effect on cell membrane biosynthesis, mediated by inhibition of the HMG- CoA reductase, there is accumulating evidence for an alternative or addi- tional mechanism of action where statins inhibit methanogenesis directly. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China
Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China Di Wu Northeast Forestry University Caihong Zhao Northeast Forestry University Hui Bai Forestry Science Research Institute of Heilongjiang Province Fujuan Feng Northeast Forestry University Xin Sui Heilongjiang University Guangyu Sun ( [email protected] ) Northeast Forestry University Research article Keywords: Wetlands, Methanogens, Community diversity, Indicator species, Methanogenic metabolic patterns Posted Date: August 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-54821/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Background: Soil methanogenic microorganisms are one of the primary methane-producing microbes in wetlands. However, we still poorly understand the community characteristic and metabolic patterns of these microorganisms according to vegetation type and seasonal changes. Therefore, to better elucidate the effects of the vegetation type and seasonal factors on the methanogenic community structure and metabolic patterns, we detected the characteristics of the soil methanogenic mcrA gene from three types of natural wetlands in different seasons in the Xiaoxing'an Mountain region, China. Result: The results indicated that the distribution of Methanobacteriaceae (hydrogenotrophic methanogens) was higher in winter, while Methanosarcinaceae and Methanosaetaceae accounted for a higher proportion in summer. Hydrogenotrophic methanogenesis was the dominant trophic pattern in each wetland. The results of principal coordinate analysis and cluster analysis showed that the vegetation type considerably inuenced the methanogenic community composition. The methanogenic community structure in the Betula platyphylla – Larix gmelinii wetland was relatively different from the structure of the other two wetland types. -

Archaeology of Eukaryotic DNA Replication
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Archaeology of Eukaryotic DNA Replication Kira S. Makarova and Eugene V. Koonin National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland 20894 Correspondence: [email protected] Recent advances in the characterization of the archaeal DNA replication system together with comparative genomic analysis have led to the identification of several previously un- characterized archaeal proteins involved in replication and currently reveal a nearly com- plete correspondence between the components of the archaeal and eukaryotic replication machineries. It can be inferred that the archaeal ancestor of eukaryotes and even the last common ancestor of all extant archaea possessed replication machineries that were compa- rable in complexity to the eukaryotic replication system. The eukaryotic replication system encompasses multiple paralogs of ancestral components such that heteromeric complexes in eukaryotes replace archaeal homomeric complexes, apparently along with subfunctionali- zation of the eukaryotic complex subunits. In the archaea, parallel, lineage-specific dupli- cations of many genes encoding replication machinery components are detectable as well; most of these archaeal paralogs remain to be functionally characterized. The archaeal rep- lication system shows remarkable plasticity whereby even some essential components such as DNA polymerase and single-stranded DNA-binding protein are displaced by unrelated proteins with analogous activities in some lineages. ouble-stranded DNA is the molecule that Okazaki fragments (Kornberg and Baker 2005; Dcarries genetic information in all cellular Barry and Bell 2006; Hamdan and Richardson life-forms; thus, replication of this genetic ma- 2009; Hamdan and van Oijen 2010). -
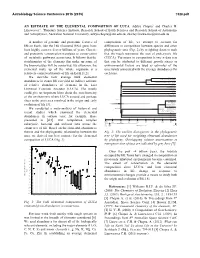
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Novel Insights Into the Thaumarchaeota in the Deepest Oceans: Their Metabolism and Potential Adaptation Mechanisms
Zhong et al. Microbiome (2020) 8:78 https://doi.org/10.1186/s40168-020-00849-2 RESEARCH Open Access Novel insights into the Thaumarchaeota in the deepest oceans: their metabolism and potential adaptation mechanisms Haohui Zhong1,2, Laura Lehtovirta-Morley3, Jiwen Liu1,2, Yanfen Zheng1, Heyu Lin1, Delei Song1, Jonathan D. Todd3, Jiwei Tian4 and Xiao-Hua Zhang1,2,5* Abstract Background: Marine Group I (MGI) Thaumarchaeota, which play key roles in the global biogeochemical cycling of nitrogen and carbon (ammonia oxidizers), thrive in the aphotic deep sea with massive populations. Recent studies have revealed that MGI Thaumarchaeota were present in the deepest part of oceans—the hadal zone (depth > 6000 m, consisting almost entirely of trenches), with the predominant phylotype being distinct from that in the “shallower” deep sea. However, little is known about the metabolism and distribution of these ammonia oxidizers in the hadal water. Results: In this study, metagenomic data were obtained from 0–10,500 m deep seawater samples from the Mariana Trench. The distribution patterns of Thaumarchaeota derived from metagenomics and 16S rRNA gene sequencing were in line with that reported in previous studies: abundance of Thaumarchaeota peaked in bathypelagic zone (depth 1000–4000 m) and the predominant clade shifted in the hadal zone. Several metagenome-assembled thaumarchaeotal genomes were recovered, including a near-complete one representing the dominant hadal phylotype of MGI. Using comparative genomics, we predict that unexpected genes involved in bioenergetics, including two distinct ATP synthase genes (predicted to be coupled with H+ and Na+ respectively), and genes horizontally transferred from other extremophiles, such as those encoding putative di-myo-inositol-phosphate (DIP) synthases, might significantly contribute to the success of this hadal clade under the extreme condition. -

Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio
International Journal of New Technology and Research (IJNTR) ISSN:2454-4116, Volume-2, Issue-9, September 2016 Pages 81-86 Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio S. Matsuda, T. Ohtsuki* Anaerobic digestion is a conventional method for biomass Abstract— Acclimation of microbial communities, aiming to utilization [8]. Despite the fact that a great deal of research methane production from grass as a sole substrate at extremely for methane production from grass has been conducted high carbon-to-nitrogen (C/N) ratio, was conducted. In a series especially in recent years, almost processes need supply of of experiments with various sizes of added grass, two microbial communities showing high methane production were obtained abundant sludge source such as sewage and manure [9, 10]. with powdered grass. In the two microbial communities Many studies indicated that the optimal carbon-to-nitrogen designated NR and RP, Bacteroidia including genera (C/N) ratio in methane fermentation were 25-30, and the C/N Bacteroides, Dysgonomonas, Proteiniphilum, and Alistipes were ratio higher than 40 are not generally suitable [11]. The C/N detected as dominant members in eubacteria. It was also shown ration of raw grass shows a wide range; wild grassy weed is at that Methanomicrobia and Methanobacteria including genera relatively high (>59) while lawn grass is at low (<29) [12]. Methanomassiliicoccus and Methanobacterium were found as dominant members in methanogen. It is noteworthy that Therefore, it is difficult to conduct methane fermentation nitrogen fixation were observed both in NR and RP, suggesting with wild grass-only, also being due to less degradability of that insufficiency of nitrogen sources would be complemented lignocellulose [13]. -

Universidad Politécnica De Madrid Escuela Técnica Superior De Ingeniería Agronómica Alimentaria Y De Biosistemas Departamento De Biotecnología-Biología Vegetal
UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL TESIS DOCTORAL Caracterización de una nueva proteína hipertermófila NifB y su papel en el mecanismo de síntesis de NifB-co, precursor del grupo metálico de FeMo-co Author: Alessandro Scandurra Director: Prof. Luis Manuel Rubio Herrero Co-director: Dr. Simon Jean Marius Arragain i ii UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL Memoria presentada por D. Alessandro Scandurra para optar al grado de Doctor Director Dr. Luis Manuel Rubio Herrero Profesor Titular UPM Madrid, 2017 © This copy oF the thesis has been supplied on condition that anyone who consults it is understood to recognize that its copyright rest with the author and that no quotation From the thesis, or any inFormation derived therefrom may be published without the author’s prior, written consent. I II AcKnowledgments As each journey, also this one has an end (Thanks goodness). It was a long trip, with several changes oF direction. Like For many other people, the PhD adventure was not easy, but I have to admit that I was lucky enough to meet really unique people, that became special Friendships. First oF all, I want to thank my Family, especially my mother and my Father, who always push me to go Forward in my life and to Follow my dreams, ready to pay the sacrifice oF the distance. This expirience has helped me to understand how easy is to be a son and how challenging is to be a parent. -

1 Development of Prediction Models of Methane Production by Sheep
Development of Prediction Models of Methane Production by Sheep and Cows Using Rumen Microbiota Data Thesis Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Boyang Zhang Graduate Program in Animal Sciences The Ohio State University 2018 Master's Examination Committee: Dr. Zhongtang Yu, Advisor Dr. Moraes, Co-advisor Dr. Firkins 1 Copyrighted by Boyang Zhang 2018 2 Abstract Methane emission from the rumen leads to approximately 10 % loss of the ingested energy, at the conversion of digestible to metabolizable energy. Besides research on mitigation of methane emission, much research interests have also been gravitated towards development of prediction models of methane emissions from livestock because the global warming is reducing agriculture productivity (Johnson and Johnson, 1995). An accurate prediction of enteric methane production from cattle and sheep can assist in balancing the increased livestock production with subsequent environmental impacts. Methane is an inevitable byproduct of the microbial fermentation processes in the rumen, and certain ruminal microbes have direct impacts to methane production (Morgavi et al., 2010). Thus, we hypothesized that the inclusion of individual microbial groups as predictor variables could improve the robustness and accuracy of prediction models. However, inclusion of microbial variables into prediction models can result in overfitting. Machine-learning algorithms can automatically select the key explanatory predictors, and Linear Mixed Models can provide a framework to predict random effects describing between animal variations. We proposed three novel frameworks for subset selections of microbial variables (MV) and one framework for generalized linear mixed models (GLMM) using L1-penalization (GLMMLASSO) selection with cross-validations (CV) ii to address the overfitting problems and to develop parsimonious prediction models of methane production. -

Lifestyle Preferences Drive the Structure and Diversity of Bacterial and Archaeal Communities in a Small Riverine Reservoir
www.nature.com/scientificreports OPEN Lifestyle preferences drive the structure and diversity of bacterial and archaeal communities in a small riverine reservoir Carles Borrego1,2, Sergi Sabater1,3* & Lorenzo Proia1,4 Spatial heterogeneity along river networks is interrupted by dams, afecting the transport, processing, and storage of organic matter, as well as the distribution of biota. We here investigated the structure of planktonic (free-living, FL), particle-attached (PA) and sediment-associated (SD) bacterial and archaeal communities within a small reservoir. We combined targeted-amplicon sequencing of bacterial and archaeal 16S rRNA genes in the DNA and RNA community fractions from FL, PA and SD, followed by imputed functional metagenomics, in order to unveil diferences in their potential metabolic capabilities within the reservoir (tail, mid, and dam sections) and lifestyles (FL, PA, SD). Both bacterial and archaeal communities were structured according to their life-style preferences rather than to their location in the reservoir. Bacterial communities were richer and more diverse when attached to particles or inhabiting the sediment, while Archaea showed an opposing trend. Diferences between PA and FL bacterial communities were consistent at functional level, the PA community showing higher potential capacity to degrade complex carbohydrates, aromatic compounds, and proteinaceous materials. Our results stressed that particle-attached prokaryotes were phylogenetically and metabolically distinct from their free-living counterparts, and that performed as hotspots for organic matter processing within the small reservoir. Spatial heterogeneity of river networks results from the sequence of lotic segments—which promote the transport and quick transformation of materials—and lentic segments such as large pools and wetlands—which mostly contribute to the process and storage of organic matter 1,2. -

Archaea and the Origin of Eukaryotes
REVIEWS Archaea and the origin of eukaryotes Laura Eme, Anja Spang, Jonathan Lombard, Courtney W. Stairs and Thijs J. G. Ettema Abstract | Woese and Fox’s 1977 paper on the discovery of the Archaea triggered a revolution in the field of evolutionary biology by showing that life was divided into not only prokaryotes and eukaryotes. Rather, they revealed that prokaryotes comprise two distinct types of organisms, the Bacteria and the Archaea. In subsequent years, molecular phylogenetic analyses indicated that eukaryotes and the Archaea represent sister groups in the tree of life. During the genomic era, it became evident that eukaryotic cells possess a mixture of archaeal and bacterial features in addition to eukaryotic-specific features. Although it has been generally accepted for some time that mitochondria descend from endosymbiotic alphaproteobacteria, the precise evolutionary relationship between eukaryotes and archaea has continued to be a subject of debate. In this Review, we outline a brief history of the changing shape of the tree of life and examine how the recent discovery of a myriad of diverse archaeal lineages has changed our understanding of the evolutionary relationships between the three domains of life and the origin of eukaryotes. Furthermore, we revisit central questions regarding the process of eukaryogenesis and discuss what can currently be inferred about the evolutionary transition from the first to the last eukaryotic common ancestor. Sister groups Two descendants that split The pioneering work by Carl Woese and colleagues In this Review, we discuss how culture- independent from the same node; the revealed that all cellular life could be divided into three genomics has transformed our understanding of descendants are each other’s major evolutionary lines (also called domains): the archaeal diversity and how this has influenced our closest relative.