WARREN LYNWOOD GARDNER Expression Vectors for the Methane-Producing Archaeon Methanococcus Maripaludis (Under the Direction of WILLIAM B
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Archaeology of Eukaryotic DNA Replication
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Archaeology of Eukaryotic DNA Replication Kira S. Makarova and Eugene V. Koonin National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland 20894 Correspondence: [email protected] Recent advances in the characterization of the archaeal DNA replication system together with comparative genomic analysis have led to the identification of several previously un- characterized archaeal proteins involved in replication and currently reveal a nearly com- plete correspondence between the components of the archaeal and eukaryotic replication machineries. It can be inferred that the archaeal ancestor of eukaryotes and even the last common ancestor of all extant archaea possessed replication machineries that were compa- rable in complexity to the eukaryotic replication system. The eukaryotic replication system encompasses multiple paralogs of ancestral components such that heteromeric complexes in eukaryotes replace archaeal homomeric complexes, apparently along with subfunctionali- zation of the eukaryotic complex subunits. In the archaea, parallel, lineage-specific dupli- cations of many genes encoding replication machinery components are detectable as well; most of these archaeal paralogs remain to be functionally characterized. The archaeal rep- lication system shows remarkable plasticity whereby even some essential components such as DNA polymerase and single-stranded DNA-binding protein are displaced by unrelated proteins with analogous activities in some lineages. ouble-stranded DNA is the molecule that Okazaki fragments (Kornberg and Baker 2005; Dcarries genetic information in all cellular Barry and Bell 2006; Hamdan and Richardson life-forms; thus, replication of this genetic ma- 2009; Hamdan and van Oijen 2010). -
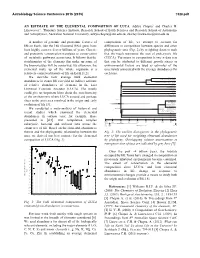
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Universidad Politécnica De Madrid Escuela Técnica Superior De Ingeniería Agronómica Alimentaria Y De Biosistemas Departamento De Biotecnología-Biología Vegetal
UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL TESIS DOCTORAL Caracterización de una nueva proteína hipertermófila NifB y su papel en el mecanismo de síntesis de NifB-co, precursor del grupo metálico de FeMo-co Author: Alessandro Scandurra Director: Prof. Luis Manuel Rubio Herrero Co-director: Dr. Simon Jean Marius Arragain i ii UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL Memoria presentada por D. Alessandro Scandurra para optar al grado de Doctor Director Dr. Luis Manuel Rubio Herrero Profesor Titular UPM Madrid, 2017 © This copy oF the thesis has been supplied on condition that anyone who consults it is understood to recognize that its copyright rest with the author and that no quotation From the thesis, or any inFormation derived therefrom may be published without the author’s prior, written consent. I II AcKnowledgments As each journey, also this one has an end (Thanks goodness). It was a long trip, with several changes oF direction. Like For many other people, the PhD adventure was not easy, but I have to admit that I was lucky enough to meet really unique people, that became special Friendships. First oF all, I want to thank my Family, especially my mother and my Father, who always push me to go Forward in my life and to Follow my dreams, ready to pay the sacrifice oF the distance. This expirience has helped me to understand how easy is to be a son and how challenging is to be a parent. -

Pan-Genome Analysis and Ancestral State Reconstruction Of
www.nature.com/scientificreports OPEN Pan‑genome analysis and ancestral state reconstruction of class halobacteria: probability of a new super‑order Sonam Gaba1,2, Abha Kumari2, Marnix Medema 3 & Rajeev Kaushik1* Halobacteria, a class of Euryarchaeota are extremely halophilic archaea that can adapt to a wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl. It consists of the orders: Halobacteriales, Haloferaciales and Natriabales. Pan‑genome analysis of class Halobacteria was done to explore the core (300) and variable components (Softcore: 998, Cloud:36531, Shell:11784). The core component revealed genes of replication, transcription, translation and repair, whereas the variable component had a major portion of environmental information processing. The pan‑gene matrix was mapped onto the core‑gene tree to fnd the ancestral (44.8%) and derived genes (55.1%) of the Last Common Ancestor of Halobacteria. A High percentage of derived genes along with presence of transformation and conjugation genes indicate the occurrence of horizontal gene transfer during the evolution of Halobacteria. A Core and pan‑gene tree were also constructed to infer a phylogeny which implicated on the new super‑order comprising of Natrialbales and Halobacteriales. Halobacteria1,2 is a class of phylum Euryarchaeota3 consisting of extremely halophilic archaea found till date and contains three orders namely Halobacteriales4,5 Haloferacales5 and Natrialbales5. Tese microorganisms are able to dwell at wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl6. Halobacteria, as the name suggests were once considered a part of a domain "Bacteria" but with the discovery of the third domain "Archaea" by Carl Woese et al.7, it became part of Archaea. -
![Downloaded from the NCBI FTP Site [37]](https://docslib.b-cdn.net/cover/4020/downloaded-from-the-ncbi-ftp-site-37-1134020.webp)
Downloaded from the NCBI FTP Site [37]
Life 2015, 5, 818-840; doi:10.3390/life5010818 OPEN ACCESS life ISSN 2075-1729 www.mdpi.com/journal/life Article Archaeal Clusters of Orthologous Genes (arCOGs): An Update and Application for Analysis of Shared Features between Thermococcales, Methanococcales, and Methanobacteriales Kira S. Makarova *, Yuri I. Wolf and Eugene V. Koonin National Center for Biotechnology Information, NLM, National Institutes of Health, Bethesda, MD 20894, USA; E-Mails: [email protected] (Y.I.W.); [email protected] (E.V.K.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-301-435-5913; Fax: +1-301-435-7793. Academic Editors: Hans-Peter Klenk, Michael W. W. Adams and Roger A. Garrett Received: 12 January 2015 / Accepted: 28 February 2015 / Published: 10 March 2015 Abstract: With the continuously accelerating genome sequencing from diverse groups of archaea and bacteria, accurate identification of gene orthology and availability of readily expandable clusters of orthologous genes are essential for the functional annotation of new genomes. We report an update of the collection of archaeal Clusters of Orthologous Genes (arCOGs) to cover, on average, 91% of the protein-coding genes in 168 archaeal genomes. The new arCOGs were constructed using refined algorithms for orthology identification combined with extensive manual curation, including incorporation of the results of several completed and ongoing research projects in archaeal genomics. A new level of classification is introduced, superclusters that unit two or more arCOGs and more completely reflect gene family evolution than individual, disconnected arCOGs. Assessment of the current archaeal genome annotation in public databases indicates that consistent use of arCOGs can significantly improve the annotation quality. -

Tackling the Methanopyrus Kandleri Paradox Céline Brochier*, Patrick Forterre† and Simonetta Gribaldo†
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central Open Access Research2004BrochieretVolume al. 5, Issue 3, Article R17 Archaeal phylogeny based on proteins of the transcription and comment translation machineries: tackling the Methanopyrus kandleri paradox Céline Brochier*, Patrick Forterre† and Simonetta Gribaldo† Addresses: *Equipe Phylogénomique, Université Aix-Marseille I, Centre Saint-Charles, 13331 Marseille Cedex 3, France. †Institut de Génétique et Microbiologie, CNRS UMR 8621, Université Paris-Sud, 91405 Orsay, France. reviews Correspondence: Céline Brochier. E-mail: [email protected] Published: 26 February 2004 Received: 14 November 2003 Revised: 5 January 2004 Genome Biology 2004, 5:R17 Accepted: 21 January 2004 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2004/5/3/R17 reports © 2004 Brochier et al.; licensee BioMed Central Ltd. This is an Open Access article: verbatim copying and redistribution of this article are permitted in all media for any purpose, provided this notice is preserved along with the article's original URL. ArchaealPhylogeneticsequencedusingrespectively). two phylogeny concatenated genomes, analysis based it of is datasetsthe now on Archaea proteinspossible consisting has ofto been thetest of transcription alternative mainly14 proteins established approach involv and translationed byes in 16S bytranscription rRNAusing machineries: largesequence andsequence 53comparison.tackling ribosomal datasets. the Methanopyrus Withproteins We theanalyzed accumulation(3,275 archaealkandleri and 6,377 of phyparadox comp positions,logenyletely Abstract deposited research Background: Phylogenetic analysis of the Archaea has been mainly established by 16S rRNA sequence comparison. With the accumulation of completely sequenced genomes, it is now possible to test alternative approaches by using large sequence datasets. -

Extremophiles
Extremophiles These microbes thrive under conditions that would kill other creatures. The molecules that enable extremophiles to prosper are becoming useful to industry by Michael T. Madigan and Barry L. Marrs DEEP-SEA VENT HEAT-LOVING MICROBES (THERMOPHILES AND HYPERTHERMOPHILES) SEA ICE COLD-LOVING MICROBES (PSYCHROPHILES) Methanopyrus kandleri Polaromonas vacuolata thereby increasing efficiency and reduc- magine diving into a refreshingly ing costs. They can also form the basis of cool swimming pool. Now, think entirely new enzyme-based processes. I instead of plowing into water that tially serve in an array of applications. Perhaps 20 research groups in the U.S., is boiling or near freezing. Or consider Of particular interest are the enzymes Japan, Germany and elsewhere are now jumping into vinegar, household am- (biological catalysts) that help extremo- actively searching for extremophiles and monia or concentrated brine. The leap philes to function in brutal circumstanc- their enzymes. Although only a few ex- would be disastrous for a person. Yet es. Like synthetic catalysts, enzymes, tremozymes have made their way into many microorganisms make their home which are proteins, speed up chemical use thus far, others are sure to follow. As in such forbidding environments. These reactions without being altered them- is true of standard enzymes, transform- microbes are called extremophiles be- selves. Last year the biomedical field and ing a newly isolated extremozyme into cause they thrive under conditions that, other industries worldwide spent more a viable product for industry can take from the human vantage, are clearly ex- than $2.5 billion on enzymes for appli- several years. -

Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities
Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book Advances in Applied Microbiology, Vol 66, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial From: Noha Youssef, Mostafa S. Elshahed, and Michael J. McInerney, Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities. In Allen I. Laskin, Sima Sariaslani, and Geoffrey M. Gadd, editors: Advances in Applied Microbiology, Vol 66, Burlington: Academic Press, 2009, pp. 141-251. ISBN: 978-0-12-374788-4 © Copyright 2009 Elsevier Inc. Academic Press. Author's personal copy CHAPTER 6 Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities Noha Youssef, Mostafa S. Elshahed, and Michael J. McInerney1 Contents I. Introduction 142 II. Factors Governing Oil Recovery 144 III. Microbial Ecology of Oil Reservoirs 147 A. Origins of microorganisms recovered from oil reservoirs 147 B. Microorganisms isolated from oil reservoirs 148 C. Culture-independent analysis of microbial communities in oil reservoirs 155 IV. -

Extremophiles — Link Between Earth and Astrobiology
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Directory of Open Access Journals Zbornik Matice srpske za prirodne nauke / Proc. Nat. Sci, Matica Srpska Novi Sad, ¥ 114, 5—16, 2008 UDC 133.52:57 Dejan B. Stojanoviã1 , Oliver O. Fojkar2 , Aleksandra V. Drobac-Åik1 , Kristina O. Åajko3 , Tamara I. Duliã1 ,ZoricaB.Sviråev1 1 Faculty of Sciences, Department of Biology and Ecology, Trg Dositeja Obradoviãa 2, 21000 Novi Sad, Serbia 2 Institute for nature conservation of Serbia, Radniåka 20A, 21000 Novi Sad, Serbia 3 Faculty of Sciences, Department of Physics, Trg Dositeja Obradoviãa 4, 21000 Novi Sad, Serbia EXTREMOPHILES — LINK BETWEEN EARTH AND ASTROBIOLOGY ABSTRACT: Astrobiology studies the origin, evolution, distribution and future of life in the universe. The most promising worlds in Solar system, beyond Earth, which may har- bor life are Mars and Jovian moon Europa. Extremophiles are organisms that thrive on the edge of temperature, hypersalinity, pH extremes, pressure, dryness and so on. In this paper, some extremophile cyanobacteria have been discussed as possible life forms in a scale of astrobiology. Samples were taken from solenetz and solonchak types of soil from the Voj- vodina region. The main idea in this paper lies in the fact that high percentage of salt found in solonchak and solonetz gives the possibility of comparison these types of soil with “soil" on Mars, which is also rich in salt. KEYWORDS: Astrobiology, extremophiles, cyanobacteria, halophiles 1. INTRODUCTION 1.1. About astrobiology Astrobiology studies the origin, evolution, distribution and future of life in the universe. -

Life in Extreme Environments
insight review articles Life in extreme environments Lynn J. Rothschild & Rocco L. Mancinelli NASA Ames Research Center, Moffett Field, California 94035-1000, USA (e-mail: [email protected]; [email protected]) Each recent report of liquid water existing elsewhere in the Solar System has reverberated through the international press and excited the imagination of humankind. Why? Because in the past few decades we have come to realize that where there is liquid water on Earth, virtually no matter what the physical conditions, there is life. What we previously thought of as insurmountable physical and chemical barriers to life, we now see as yet another niche harbouring ‘extremophiles’. This realization, coupled with new data on the survival of microbes in the space environment and modelling of the potential for transfer of life between celestial bodies, suggests that life could be more common than previously thought. Here we examine critically what it means to be an extremophile, and the implications of this for evolution, biotechnology and especially the search for life in the Universe. ormal is passé; extreme is chic. While thriving in biological extremes (for example, nutritional Aristotle cautioned “everything in extremes, and extremes of population density, parasites, moderation”, the Romans, known for their prey, and so on). excesses, coined the word ‘extremus’, the ‘Extremophile’ conjures up images of prokaryotes, yet the superlative of exter (‘being on the outside’). taxonomic range spans all three domains. Although all NBy the fifteenth century ‘extreme’ had arrived, via Middle hyperthermophiles are members of the Archaea and French, to English. At the dawning of the twenty-first Bacteria, eukaryotes are common among the psychrophiles, century we know that the Solar System, and even Earth, acidophiles, alkaliphiles, piezophiles, xerophiles and contain environmental extremes unimaginable to the halophiles (which respectively thrive at low temperatures, low ‘ancients’ of the nineteenth century.