Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Review Article: Inhibition of Methanogenic Archaea by Statins As a Targeted Management Strategy for Constipation and Related Disorders
Alimentary Pharmacology and Therapeutics Review article: inhibition of methanogenic archaea by statins as a targeted management strategy for constipation and related disorders K. Gottlieb*, V. Wacher*, J. Sliman* & M. Pimentel† *Synthetic Biologics, Inc., Rockville, SUMMARY MD, USA. † Gastroenterology, Cedars-Sinai Background Medical Center, Los Angeles, CA, USA. Observational studies show a strong association between delayed intestinal transit and the production of methane. Experimental data suggest a direct inhibitory activity of methane on the colonic and ileal smooth muscle and Correspondence to: a possible role for methane as a gasotransmitter. Archaea are the only con- Dr K. Gottlieb, Synthetic Biologics, fi Inc., 9605 Medical Center Drive, rmed biological sources of methane in nature and Methanobrevibacter Rockville, MD 20850, USA. smithii is the predominant methanogen in the human intestine. E-mail: [email protected] Aim To review the biosynthesis and composition of archaeal cell membranes, Publication data archaeal methanogenesis and the mechanism of action of statins in this context. Submitted 8 September 2015 First decision 29 September 2015 Methods Resubmitted 7 October 2015 Narrative review of the literature. Resubmitted 20 October 2015 Accepted 20 October 2015 Results EV Pub Online 11 November 2015 Statins can inhibit archaeal cell membrane biosynthesis without affecting This uncommissioned review article was bacterial numbers as demonstrated in livestock and humans. This opens subject to full peer-review. the possibility of a therapeutic intervention that targets a specific aetiologi- cal factor of constipation while protecting the intestinal microbiome. While it is generally believed that statins inhibit methane production via their effect on cell membrane biosynthesis, mediated by inhibition of the HMG- CoA reductase, there is accumulating evidence for an alternative or addi- tional mechanism of action where statins inhibit methanogenesis directly. -

The Vertical Distribution of Sediment Archaeal Community in the (Black Bloom) Disturbing Zhushan Bay of Lake Taihu
Hindawi Publishing Corporation Archaea Volume 2016, Article ID 8232135, 8 pages http://dx.doi.org/10.1155/2016/8232135 Research Article The Vertical Distribution of Sediment Archaeal Community in the (Black Bloom) Disturbing Zhushan Bay of Lake Taihu Xianfang Fan1,2 and Peng Xing1 1 State Key Laboratory of Lake Science and Environment, Nanjing Institute of Geography & Limnology, Chinese Academy of Sciences, Nanjing 210008, China 2State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing 210008, China Correspondence should be addressed to Peng Xing; [email protected] Received 20 August 2015; Revised 27 November 2015; Accepted 20 December 2015 Academic Editor: William B. Whitman Copyright © 2016 X. Fan and P. Xing. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Using the Illumina sequencing technology, we investigated the vertical distribution of archaeal community in the sediment of Zhushan Bay of Lake Taihu, where the black bloom frequently occurred in summer. Overall, the Miscellaneous Crenarchaeotal Group (MCG), Deep Sea Hydrothermal Vent Group 6 (DHVEG-6), and Methanobacterium dominated the archaeal community. However, we observed significant difference in composition of archaeal community among different depths of the sediment. DHVEG-6 dominated in the surface layer (0–3 cm) sediment. Methanobacterium was the dominating archaeal taxa in the L2 (3– 6 cm) and L3 (6–10) sediment. MCG was most abundant in the L4 (10–15 cm) and L5 (15–20 cm) sediment. Besides, DHVEG-6 was significantly affected by the concentration of total phosphorus ).(TP And loss on ignition (LOI) was an important environmental factor for Methanobacterium. -

Growth of Sedimentary Bathyarchaeota on Lignin As an Energy Source
Growth of sedimentary Bathyarchaeota on lignin as an energy source Tiantian Yua,b,1, Weichao Wuc,d,1, Wenyue Lianga,b, Mark Alexander Levere, Kai-Uwe Hinrichsc,d, and Fengping Wanga,b,2 aState Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, 200240 Shanghai, China; bState Key Laboratory of Ocean Engineering, Shanghai Jiao Tong University, 200240 Shanghai, China; cOrganic Geochemistry Group, MARUM-Center for Marine Environmental Sciences, University of Bremen, 28359 Bremen, Germany; dDepartment of Geosciences, University of Bremen, 28359 Bremen, Germany; and eInstitute of Biogeochemistry and Pollutant Dynamics, Department of Environmental Systems Science, Swiss Federal Institute of Technology Zurich, 8092 Zurich, Switzerland Edited by Edward F. DeLong, University of Hawaii at Manoa, Honolulu, HI, and approved April 16, 2018 (received for review October 30, 2017) Members of the archaeal phylum Bathyarchaeota are among the other hand, acetogenesis from H2/CO2 has been proposed for most abundant microorganisms on Earth. Although versatile met- some lineages of Bathyarchaeota (3). The reductive acetyl-CoA abolic capabilities such as acetogenesis, methanogenesis, and fer- pathway for carbon fixation has been identified in most of the mentation have been suggested for bathyarchaeotal members, no obtained bathyarchaeotal genomes (3, 12, 14). Among these, direct confirmation of these metabolic functions has been achieved members of the bathyarchaeotal subgroups Bathy-3 and Bathy- through growth of Bathyarchaeota in the laboratory. Here we dem- 8 have been suggested to be capable of methanogenesis (12). All onstrate, on the basis of gene-copy numbers and probing of ar- the above studies indicate great versatility in the metabolic po- chaeal lipids, the growth of Bathyarchaeota subgroup Bathy-8 in tentials of Bathyarchaeota, with some lineages possibly being enrichments of estuarine sediments with the biopolymer lignin. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

THE MASS of L-PYRROLYSINE in METHYLAMINE METHYLTRANSFERASES and the ROLE of ITS IMINE BOND in CATALYSIS DISSERTATION Presented I
THE MASS OF L-PYRROLYSINE IN METHYLAMINE METHYLTRANSFERASES AND THE ROLE OF ITS IMINE BOND IN CATALYSIS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Graduate School of The Ohio State University by Jitesh Anthony Aloysius Soares, M.S. The Ohio State University 2008 Dissertation Committee: Dr. Joseph A. Krzycki, Advisor Approved by Dr. Charles J. Daniels Dr. Mark Morrison ______________________ Dr. F. Robert Tabita Advisor Graduate Program in Microbiology ABSTRACT Methanosarcina barkeri is an archaeon capable of producing methane from methylamines. Methylamine methyltransferases initiate methanogenesis from methylamines by transferring methyl groups to a cognate corrinoid protein. Each gene encoding a methylamine methyltransferase has been shown to contain a single in-frame amber codon. Further studies have shown that in the monomethylamine methyltransferase, mtmB , the amber codon encodes a novel amino acid, L-pyrrolysine. X-ray crystal structures of MtmB have shown that the structure of this amino acid is a lysine residue with the epsilon-nitrogen in amide linkage to a (4R, 5R)-4-substituted pyrrolyine-5-carboxylate ring. However, these structures did not allow an assignment of the pyrroline ring C4 substituent as a methyl or amine group. In this thesis (Chapter 2) mass spectrometry of chymotryptic digests of methylamine methyltransferases is employed to show that pyrrolysine in present in all three types of methylamine methyltransferase at the position corresponding to the amber codon in their respective genes. The mass of this amber-encoded residue was observed to coincide with the predicted mass of pyrrolysine with a methyl- group at the C4 position. -

Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma Divulgatum
microorganisms Article Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum Rafael Bargiela 1 , Karin Lanthaler 1,2, Colin M. Potter 1,2 , Manuel Ferrer 3 , Alexander F. Yakunin 1,2, Bela Paizs 1,2, Peter N. Golyshin 1,2 and Olga V. Golyshina 1,2,* 1 School of Natural Sciences, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK; [email protected] (R.B.); [email protected] (K.L.); [email protected] (C.M.P.); [email protected] (A.F.Y.); [email protected] (B.P.); [email protected] (P.N.G.) 2 Centre for Environmental Biotechnology, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK 3 Systems Biotechnology Group, Department of Applied Biocatalysis, CSIC—Institute of Catalysis, Marie Curie 2, 28049 Madrid, Spain; [email protected] * Correspondence: [email protected]; Tel.: +44-1248-388607; Fax: +44-1248-382569 Received: 27 April 2020; Accepted: 15 May 2020; Published: 19 May 2020 Abstract: The archaeon Cuniculiplasma divulgatum is ubiquitous in acidic environments with low-to-moderate temperatures. However, molecular mechanisms underlying its ability to thrive at lower temperatures remain unexplored. Using mass spectrometry (MS)-based proteomics, we analysed the effect of short-term (3 h) exposure to cold. The C. divulgatum genome encodes 2016 protein-coding genes, from which 819 proteins were identified in the cells grown under optimal conditions. In line with the peptidolytic lifestyle of C. divulgatum, its intracellular proteome revealed the abundance of proteases, ABC transporters and cytochrome C oxidase. From 747 quantifiable polypeptides, the levels of 582 proteins showed no change after the cold shock, whereas 104 proteins were upregulated suggesting that they might be contributing to cold adaptation. -
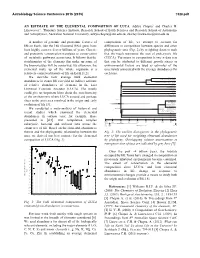
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio
International Journal of New Technology and Research (IJNTR) ISSN:2454-4116, Volume-2, Issue-9, September 2016 Pages 81-86 Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio S. Matsuda, T. Ohtsuki* Anaerobic digestion is a conventional method for biomass Abstract— Acclimation of microbial communities, aiming to utilization [8]. Despite the fact that a great deal of research methane production from grass as a sole substrate at extremely for methane production from grass has been conducted high carbon-to-nitrogen (C/N) ratio, was conducted. In a series especially in recent years, almost processes need supply of of experiments with various sizes of added grass, two microbial communities showing high methane production were obtained abundant sludge source such as sewage and manure [9, 10]. with powdered grass. In the two microbial communities Many studies indicated that the optimal carbon-to-nitrogen designated NR and RP, Bacteroidia including genera (C/N) ratio in methane fermentation were 25-30, and the C/N Bacteroides, Dysgonomonas, Proteiniphilum, and Alistipes were ratio higher than 40 are not generally suitable [11]. The C/N detected as dominant members in eubacteria. It was also shown ration of raw grass shows a wide range; wild grassy weed is at that Methanomicrobia and Methanobacteria including genera relatively high (>59) while lawn grass is at low (<29) [12]. Methanomassiliicoccus and Methanobacterium were found as dominant members in methanogen. It is noteworthy that Therefore, it is difficult to conduct methane fermentation nitrogen fixation were observed both in NR and RP, suggesting with wild grass-only, also being due to less degradability of that insufficiency of nitrogen sources would be complemented lignocellulose [13]. -

Translation of the Amber Codon in Methylamine Methyltransferase Genes of a Methanogenic Archaeon
TRANSLATION OF THE AMBER CODON IN METHYLAMINE METHYLTRANSFERASE GENES OF A METHANOGENIC ARCHAEON DISSERTATION Presented in Partial Fulfillment of the Requirements for The Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Gayathri Srinivasan, M. S. * * * * * The Ohio State University 2003 Dissertation Committee: Dr. Joseph A. Krzycki, Advisor Approved by Dr. Charles J. Daniels Dr. Tina M. Henkin ________________________ (Advisor) Dr. John N. Reeve Department of Microbiology ABSTRACT Members of the Methanosarcinaceae family can in addition to hydrogen/carbon dioxide utilize several methylated compounds and convert them to methane. Methanogenesis from methylamines involves methylamine specific methyltransferases that transfer the methyl group from the methylamines to a corrinoid protein. The methylamine specific methyltransferase genes contain a single in-frame amber codon that is not read as a translational stop. The residue encoded by the amber codon, has been found to be a novel amino acid, pyrrolysine in MtmB. Multiple copies of monomethylamine methyltransferase genes (mtmB) containing a single amber codon within their open reading frames, along with the genes encoding their cognate corrinoid proteins (mtmC), exist within the genomes of the members of the Methanosarcinaceae family. The two copies of mtmCB genes from M. barkeri MS are differentially transcribed. Editing of the mtmB2 transcript was not detected suggesting a mechanism of amber codon readthrough occurring in the organism. Similar to selenocysteine incorporation at UGA codons, the Methanosarcinaceae also appear to have a unique mechanism for amber codon readthrough. An amber decoding tRNA gene, pylT, along with its cognate lysyl tRNA synthetase, pylS, are found near the MMA methyltransferase gene cluster. -

A Systematic Approach Re-Analyzing the Effects of Temperature
www.nature.com/scientificreports OPEN A systematic approach re-analyzing the efects of temperature disturbance on the microbial Received: 12 September 2018 Accepted: 12 April 2019 community of mesophilic anaerobic Published: xx xx xxxx digestion Grace Tzun-Wen Shaw , Chieh-Yin Weng, Cheng-Yu Chen , Francis Cheng-Hsuan Weng & Daryi Wang Microbial communities are key drivers of ecosystem processes, but their behavior in disturbed environments is difcult to measure. How microbial community composition and function respond disturbances is a common challenge in biomedical, environmental, agricultural, and bioenergy research. A novel way to solve this problem is to use a systems-level perspective and describe microbial communities as networks. Based on a mesophilic anaerobic digestion system of swine manure as a tool, we propose a simple framework to investigate changes in microbial communities via compositions, metabolic pathways, genomic properties and interspecies relationships in response to a long-term temperature disturbance. After temperature disturbance, microbial communities tend towards a competitive interaction network with higher GC content and larger genome size. Based on microbial interaction networks, communities responded to the disturbance by showing a transition from acetotrophic (Methanotrichaceae and Methanosarcinaceae) to methylotrophic methanogens (Methanomassiliicoccaceae and Methanobacteriaceae) and a fuctuation in rare biosphere taxa. To conclude, this study may be important for exploring the dynamic relationships between disturbance and microbial communities as a whole, as well as for providing researchers with a better understanding of how changes in microbial communities relate to ecological processes. Environmental disturbances are important factors in shaping the structure of plant, animal or microbial pop- ulations. In the past few decades, remarkable progress has been made towards understanding how disturbance afects microbial composition, biodiversity loss and ecological processes1–5. -

1 Development of Prediction Models of Methane Production by Sheep
Development of Prediction Models of Methane Production by Sheep and Cows Using Rumen Microbiota Data Thesis Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Boyang Zhang Graduate Program in Animal Sciences The Ohio State University 2018 Master's Examination Committee: Dr. Zhongtang Yu, Advisor Dr. Moraes, Co-advisor Dr. Firkins 1 Copyrighted by Boyang Zhang 2018 2 Abstract Methane emission from the rumen leads to approximately 10 % loss of the ingested energy, at the conversion of digestible to metabolizable energy. Besides research on mitigation of methane emission, much research interests have also been gravitated towards development of prediction models of methane emissions from livestock because the global warming is reducing agriculture productivity (Johnson and Johnson, 1995). An accurate prediction of enteric methane production from cattle and sheep can assist in balancing the increased livestock production with subsequent environmental impacts. Methane is an inevitable byproduct of the microbial fermentation processes in the rumen, and certain ruminal microbes have direct impacts to methane production (Morgavi et al., 2010). Thus, we hypothesized that the inclusion of individual microbial groups as predictor variables could improve the robustness and accuracy of prediction models. However, inclusion of microbial variables into prediction models can result in overfitting. Machine-learning algorithms can automatically select the key explanatory predictors, and Linear Mixed Models can provide a framework to predict random effects describing between animal variations. We proposed three novel frameworks for subset selections of microbial variables (MV) and one framework for generalized linear mixed models (GLMM) using L1-penalization (GLMMLASSO) selection with cross-validations (CV) ii to address the overfitting problems and to develop parsimonious prediction models of methane production. -

Lifestyle Preferences Drive the Structure and Diversity of Bacterial and Archaeal Communities in a Small Riverine Reservoir
www.nature.com/scientificreports OPEN Lifestyle preferences drive the structure and diversity of bacterial and archaeal communities in a small riverine reservoir Carles Borrego1,2, Sergi Sabater1,3* & Lorenzo Proia1,4 Spatial heterogeneity along river networks is interrupted by dams, afecting the transport, processing, and storage of organic matter, as well as the distribution of biota. We here investigated the structure of planktonic (free-living, FL), particle-attached (PA) and sediment-associated (SD) bacterial and archaeal communities within a small reservoir. We combined targeted-amplicon sequencing of bacterial and archaeal 16S rRNA genes in the DNA and RNA community fractions from FL, PA and SD, followed by imputed functional metagenomics, in order to unveil diferences in their potential metabolic capabilities within the reservoir (tail, mid, and dam sections) and lifestyles (FL, PA, SD). Both bacterial and archaeal communities were structured according to their life-style preferences rather than to their location in the reservoir. Bacterial communities were richer and more diverse when attached to particles or inhabiting the sediment, while Archaea showed an opposing trend. Diferences between PA and FL bacterial communities were consistent at functional level, the PA community showing higher potential capacity to degrade complex carbohydrates, aromatic compounds, and proteinaceous materials. Our results stressed that particle-attached prokaryotes were phylogenetically and metabolically distinct from their free-living counterparts, and that performed as hotspots for organic matter processing within the small reservoir. Spatial heterogeneity of river networks results from the sequence of lotic segments—which promote the transport and quick transformation of materials—and lentic segments such as large pools and wetlands—which mostly contribute to the process and storage of organic matter 1,2.