Growth of Sedimentary Bathyarchaeota on Lignin As an Energy Source
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China
Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China Di Wu Northeast Forestry University Caihong Zhao Northeast Forestry University Hui Bai Forestry Science Research Institute of Heilongjiang Province Fujuan Feng Northeast Forestry University Xin Sui Heilongjiang University Guangyu Sun ( [email protected] ) Northeast Forestry University Research article Keywords: Wetlands, Methanogens, Community diversity, Indicator species, Methanogenic metabolic patterns Posted Date: August 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-54821/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Background: Soil methanogenic microorganisms are one of the primary methane-producing microbes in wetlands. However, we still poorly understand the community characteristic and metabolic patterns of these microorganisms according to vegetation type and seasonal changes. Therefore, to better elucidate the effects of the vegetation type and seasonal factors on the methanogenic community structure and metabolic patterns, we detected the characteristics of the soil methanogenic mcrA gene from three types of natural wetlands in different seasons in the Xiaoxing'an Mountain region, China. Result: The results indicated that the distribution of Methanobacteriaceae (hydrogenotrophic methanogens) was higher in winter, while Methanosarcinaceae and Methanosaetaceae accounted for a higher proportion in summer. Hydrogenotrophic methanogenesis was the dominant trophic pattern in each wetland. The results of principal coordinate analysis and cluster analysis showed that the vegetation type considerably inuenced the methanogenic community composition. The methanogenic community structure in the Betula platyphylla – Larix gmelinii wetland was relatively different from the structure of the other two wetland types. -

Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma Divulgatum
microorganisms Article Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum Rafael Bargiela 1 , Karin Lanthaler 1,2, Colin M. Potter 1,2 , Manuel Ferrer 3 , Alexander F. Yakunin 1,2, Bela Paizs 1,2, Peter N. Golyshin 1,2 and Olga V. Golyshina 1,2,* 1 School of Natural Sciences, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK; [email protected] (R.B.); [email protected] (K.L.); [email protected] (C.M.P.); [email protected] (A.F.Y.); [email protected] (B.P.); [email protected] (P.N.G.) 2 Centre for Environmental Biotechnology, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK 3 Systems Biotechnology Group, Department of Applied Biocatalysis, CSIC—Institute of Catalysis, Marie Curie 2, 28049 Madrid, Spain; [email protected] * Correspondence: [email protected]; Tel.: +44-1248-388607; Fax: +44-1248-382569 Received: 27 April 2020; Accepted: 15 May 2020; Published: 19 May 2020 Abstract: The archaeon Cuniculiplasma divulgatum is ubiquitous in acidic environments with low-to-moderate temperatures. However, molecular mechanisms underlying its ability to thrive at lower temperatures remain unexplored. Using mass spectrometry (MS)-based proteomics, we analysed the effect of short-term (3 h) exposure to cold. The C. divulgatum genome encodes 2016 protein-coding genes, from which 819 proteins were identified in the cells grown under optimal conditions. In line with the peptidolytic lifestyle of C. divulgatum, its intracellular proteome revealed the abundance of proteases, ABC transporters and cytochrome C oxidase. From 747 quantifiable polypeptides, the levels of 582 proteins showed no change after the cold shock, whereas 104 proteins were upregulated suggesting that they might be contributing to cold adaptation. -
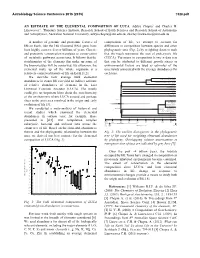
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Pan-Genome Analysis and Ancestral State Reconstruction Of
www.nature.com/scientificreports OPEN Pan‑genome analysis and ancestral state reconstruction of class halobacteria: probability of a new super‑order Sonam Gaba1,2, Abha Kumari2, Marnix Medema 3 & Rajeev Kaushik1* Halobacteria, a class of Euryarchaeota are extremely halophilic archaea that can adapt to a wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl. It consists of the orders: Halobacteriales, Haloferaciales and Natriabales. Pan‑genome analysis of class Halobacteria was done to explore the core (300) and variable components (Softcore: 998, Cloud:36531, Shell:11784). The core component revealed genes of replication, transcription, translation and repair, whereas the variable component had a major portion of environmental information processing. The pan‑gene matrix was mapped onto the core‑gene tree to fnd the ancestral (44.8%) and derived genes (55.1%) of the Last Common Ancestor of Halobacteria. A High percentage of derived genes along with presence of transformation and conjugation genes indicate the occurrence of horizontal gene transfer during the evolution of Halobacteria. A Core and pan‑gene tree were also constructed to infer a phylogeny which implicated on the new super‑order comprising of Natrialbales and Halobacteriales. Halobacteria1,2 is a class of phylum Euryarchaeota3 consisting of extremely halophilic archaea found till date and contains three orders namely Halobacteriales4,5 Haloferacales5 and Natrialbales5. Tese microorganisms are able to dwell at wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl6. Halobacteria, as the name suggests were once considered a part of a domain "Bacteria" but with the discovery of the third domain "Archaea" by Carl Woese et al.7, it became part of Archaea. -

Phylogeny and Taxonomy of Archaea: a Comparison of the Whole-Genome-Based Cvtree Approach with 16S Rrna Sequence Analysis
Life 2015, 5, 949-968; doi:10.3390/life5010949 OPEN ACCESS life ISSN 2075-1729 www.mdpi.com/journal/life Article Phylogeny and Taxonomy of Archaea: A Comparison of the Whole-Genome-Based CVTree Approach with 16S rRNA Sequence Analysis Guanghong Zuo 1, Zhao Xu 2 and Bailin Hao 1;* 1 T-Life Research Center and Department of Physics, Fudan University, 220 Handan Road, Shanghai 200433, China; E-Mail: [email protected] 2 Thermo Fisher Scientific, 200 Oyster Point Blvd, South San Francisco, CA 94080, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +86-21-6565-2305. Academic Editors: Roger A. Garrett, Hans-Peter Klenk and Michael W. W. Adams Received: 9 December 2014 / Accepted: 9 March 2015 / Published: 17 March 2015 Abstract: A tripartite comparison of Archaea phylogeny and taxonomy at and above the rank order is reported: (1) the whole-genome-based and alignment-free CVTree using 179 genomes; (2) the 16S rRNA analysis exemplified by the All-Species Living Tree with 366 archaeal sequences; and (3) the Second Edition of Bergey’s Manual of Systematic Bacteriology complemented by some current literature. A high degree of agreement is reached at these ranks. From the newly proposed archaeal phyla, Korarchaeota, Thaumarchaeota, Nanoarchaeota and Aigarchaeota, to the recent suggestion to divide the class Halobacteria into three orders, all gain substantial support from CVTree. In addition, the CVTree helped to determine the taxonomic position of some newly sequenced genomes without proper lineage information. A few discrepancies between the CVTree and the 16S rRNA approaches call for further investigation. -

Article Mode, Which Adds to a Better and Living Conditions of Microorganisms in the Atmosphere Understanding of the Overall Atmospheric Microbiome
Biogeosciences, 15, 4205–4214, 2018 https://doi.org/10.5194/bg-15-4205-2018 © Author(s) 2018. This work is distributed under the Creative Commons Attribution 4.0 License. Community composition and seasonal changes of archaea in coarse and fine air particulate matter Jörn Wehking1,2, Daniel A. Pickersgill1,2, Robert M. Bowers3,4, David Teschner1,2, Ulrich Pöschl2, Janine Fröhlich-Nowoisky2, and Viviane R. Després1,2 1Institute of Molecular Physiology, Johannes Gutenberg University, Johannes-von-Müller-Weg 6, 55128 Mainz, Germany 2Max Planck Institute for Chemistry, P.O. Box 3060, 55020 Mainz, Germany 3DOE Joint Genome Institute, Walnut Creek, CA, USA 4University of Colorado, Ecology and Evolutionary Biology, Boulder, CO, USA Correspondence: Viviane R. Després ([email protected]) and Jörn Wehking ([email protected]) Received: 1 December 2017 – Discussion started: 14 December 2017 Revised: 11 June 2018 – Accepted: 15 June 2018 – Published: 11 July 2018 Abstract. Archaea are ubiquitous in terrestrial and marine narchaeaceae, Nitrososphaeraceae, Methanosarcinales, Ther- environments and play an important role in biogeochemical moplasmata, and the genus Nitrosopumilus as the dominating cycles. Although air acts as the primary medium for their taxa. dispersal among different habitats, their diversity and abun- The seasonal dynamics of methanogenic Euryarchaeota dance is not well characterized. The main reason for this point to anthropogenic activities, such as fertilization of agri- lack of insight is that archaea are difficult to culture, seem cultural fields with biogas substrates or manure, as sources of to be low in number in the atmosphere, and have so far been airborne archaea. This study gains a deeper insight into the difficult to detect even with molecular genetic approaches. -

Salinity Drives Archaeal Distribution Patterns in High Altitude Lake Sediments on the Tibetan Plateau Yongqin Liu 1,2,∗, John C
FEMS Microbiology Ecology, 92, 2016, fiw033 doi: 10.1093/femsec/fiw033 Advance Access Publication Date: 16 February 2016 Research Article RESEARCH ARTICLE Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau Yongqin Liu 1,2,∗, John C. Priscu3, Jinbo Xiong4, Ralf Conrad5, Trista Vick-Majors3, Haiyan Chu6 and Juzhi Hou1 Downloaded from 1Key Laboratory of Tibetan Environment Changes and Land Surface Processes, Institute of Tibetan Plateau Research, Beijing 100101, China, 2CAS Center for Excellence in Tibetan Plateau Earth Sciences, Chinese Academy of Sciences, Beijing 100101, China, 3Department of Land Resources and Environmental Sciences, Montana State University, Bozeman, MT 59717, USA, 4Faculty of Marine Sciences, Ningbo University, Ningbo http://femsec.oxfordjournals.org/ 315211, China, 5Max Planck Institute for Terrestrial Microbiology, Marburg 35043, Germany and 6State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing 210008, China ∗Corresponding author: Institute of Tibetan Plateau, Chinese Academy of Science, No. 16, Lincuo Rd., Beijing 100101, China. Tel: 86-10-84097122; E-mail: [email protected] One sentence summary: We represent a comprehensive investigation of archaeal diversity in the pristine lake sediments across the Tibetan Plateau, and found salinity is the key factor controlling archaeal community diversity and composition. Editor: Dirk Wagner by guest on April 5, 2016 ABSTRACT Archaeal communities and the factors regulating their diversity in high altitude lakes are poorly understood. Here, we provide the first high-throughput sequencing study of Archaea from Tibetan Plateau lake sediments. We analyzed twenty lake sediments from the world’s highest and largest plateau and found diverse archaeal assemblages that clustered into groups dominated by methanogenic Euryarchaeota, Crenarchaeota and Halobacteria/mixed euryarchaeal phylotypes. -

Distribution and Evolution of the Mobile Vma-1B Intein
Authors' pre-print version. Manuscript published in Molecular Biology and Evolution MBE-13-0254.R2 DOI of published manuscript: 10.1093/molbev/mst164 Article Discoveries Section Distribution and Evolution of the Mobile vma-1b Intein. Kristen S. Swithers1,3, Shannon M. Soucy1, Erica Lasek-Nesselquist1,5, Pascal Lapierre2,4 and Johann Peter Gogarten1* 1 Department of Molecular and Cell Biology, University of Connecticut, Storrs, Connecticut, United States of America 2 University of Connecticut Biotechnology Center, University of Connecticut, Storrs, Connecticut, United States of America 3 Current affiliation: Department of Cell Biology, Yale University Medical School, New Haven, Connecticut, United States of America 4 Current affiliation: New York State Department of Health, Wadsworth Center, Albany, New York, United States of America 5 Current affiliation: Department of Biology, University of Scranton, Scranton, PA, United States of America Communicating author: Johann Peter Gogarten Communicating author email: [email protected], [email protected] !"#"!" Abstract Inteins are self-splicing parasitic genetic elements found in all domains of life. These genetic elements are found in highly conserved positions in conserved proteins. One protein family that has been invaded by inteins is the vacuolar and archaeal catalytic ATPase subunits (vma-1). There are two intein insertion sites in this protein, "a" and "b". The "b" site was previously thought to be only invaded in archaeal lineages. Here we survey the distribution and evolutionary histories of the "b" site inteins and show the intein is present in more lineages than previously annotated including a bacterial lineage, Mahalla australienses 50-1 BON. We present evidence, through ancestral character state reconstruction and substitution ratios between host genes and inteins, for several transfers of this intein between divergent species, including an interdomain transfer between the Archaea and Bacteria. -

Variations in the Two Last Steps of the Purine Biosynthetic Pathway in Prokaryotes
GBE Different Ways of Doing the Same: Variations in the Two Last Steps of the Purine Biosynthetic Pathway in Prokaryotes Dennifier Costa Brandao~ Cruz1, Lenon Lima Santana1, Alexandre Siqueira Guedes2, Jorge Teodoro de Souza3,*, and Phellippe Arthur Santos Marbach1,* 1CCAAB, Biological Sciences, Recoˆ ncavo da Bahia Federal University, Cruz das Almas, Bahia, Brazil 2Agronomy School, Federal University of Goias, Goiania,^ Goias, Brazil 3 Department of Phytopathology, Federal University of Lavras, Minas Gerais, Brazil Downloaded from https://academic.oup.com/gbe/article/11/4/1235/5345563 by guest on 27 September 2021 *Corresponding authors: E-mails: [email protected]fla.br; [email protected]. Accepted: February 16, 2019 Abstract The last two steps of the purine biosynthetic pathway may be catalyzed by different enzymes in prokaryotes. The genes that encode these enzymes include homologs of purH, purP, purO and those encoding the AICARFT and IMPCH domains of PurH, here named purV and purJ, respectively. In Bacteria, these reactions are mainly catalyzed by the domains AICARFT and IMPCH of PurH. In Archaea, these reactions may be carried out by PurH and also by PurP and PurO, both considered signatures of this domain and analogous to the AICARFT and IMPCH domains of PurH, respectively. These genes were searched for in 1,403 completely sequenced prokaryotic genomes publicly available. Our analyses revealed taxonomic patterns for the distribution of these genes and anticorrelations in their occurrence. The analyses of bacterial genomes revealed the existence of genes coding for PurV, PurJ, and PurO, which may no longer be considered signatures of the domain Archaea. Although highly divergent, the PurOs of Archaea and Bacteria show a high level of conservation in the amino acids of the active sites of the protein, allowing us to infer that these enzymes are analogs. -

Methanocaldococcus Jannaschii Mja Methanococci Methanococcales
Table S2 List of the archaeal species used for evaluation of physiological and metabolic potential by MAPLE system Species Species ID Class Order [Euryarchaeota: 53 species] Methanocaldococcus jannaschii mja Methanococci Methanococcales Methanotorris igneus mig Methanococci Methanococcales Methanococcus vannielii mvn Methanococci Methanococcales Methanothermococcus okinawensis mok Methanococci Methanococcales Methanosarcina barkeri mba Methanomicrobia Methanosarcinales Methanococcoides burtonii mbu Methanomicrobia Methanosarcinales Methanohalophilus mahii mmh Methanomicrobia Methanosarcinales Methanohalobium evestigatum mev Methanomicrobia Methanosarcinales Methanosalsum zhilinae mzh Methanomicrobia Methanosarcinales Methanolobus psychrophilus mpy Methanomicrobia Methanosarcinales Methanomethylovorans hollandica mhz Methanomicrobia Methanosarcinales Methanosaeta concilii mcj Methanomicrobia Methanosarcinales Methanospirillum hungatei mhu Methanomicrobia Methanomicrobiales Methanocorpusculum labreanum mla Methanomicrobia Methanomicrobiales Methanoculleus bourgensis mbg Methanomicrobia Methanomicrobiales Methanoplanus petrolearius mpi Methanomicrobia Methanomicrobiales Methanoregula boonei mbn Methanomicrobia Methanomicrobiales Candidatus Methanosphaerula palustris mpl Methanomicrobia Methanomicrobiales Methanocella paludicola mpd Methanomicrobia Methanocellales Methanomassiliicoccus sp. Mx1-Issoire mer Methanomicrobia Unclassified Methanothermobacter thermautotrophicus mth Methanobacteria Methanobacteriales Methanosphaera stadtmanae mst -

WHAT CAN LIFE on EARTH TELL US ABOUT LIFE in the UNIVERSE? CHARLES H. LINEWEAVER & ADITYA CHOPRA Planetary Science Institut
J. Seckbach (ed.), Genesis - In The Beginning: Precursors of Life, Chemical Models and Early Biological Evolution, Cellular Origin, Life in Extreme Habitats and Astrobiology 22, 799–815 DOI 10.1007/978-94-007-2941-4_40, ISBN 978-94-007-2940-7.© Springer 2012 WHAT CAN LIFE ON EARTH TELL US ABOUT LIFE IN THE UNIVERSE? CHARLES H. LINEWEAVER & ADITYA CHOPRA Planetary Science Institute, Research School of Astronomy and Astrophysics and the Research School of Earth Sciences, Australian National University, Canberra, Australia We review the most fundamental features common to all terrestrial life. We argue that the ubiquity of these features makes them the best candidates for being features of extraterrestrial life. Other frequently espoused candidates are less secure because they are based on subjective notions of universal fitness, not on features common to all terrestrial life. For example, major transitions in the evolutionary pathway that led to Homo sapiens are sometimes considered to be fundamental transitions in the evolution of all life. However, these “major transitions” are largely arbitrary because a series of different major transitions can be identified along the evolutionary pathway to any extant species. 1. Quirkometry and Terrestrial Life as a Model Organism The universe is filled with stars similar to our Sun (Robles et al., 2008), rocky planets similar to our Earth (Lineweaver and Grether, 2003; Ida and Lin 2004; Mordasini et al., 2009), water like our oceans (Kuchner, 2003; Léger et al., 2004), amino acids like our proteins and all the other ingredients for life (Pizzarello, 2007). But is the universe filled with life? If it is, what kind of life is it? We argue that if there is life out there at all, its basic features are likely to be a subset of the features common to all terrestrial life.