I EXPRESSION PROFILING of THERMOPLASMA VOLCANIUM
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Developing a Genetic Manipulation System for the Antarctic Archaeon, Halorubrum Lacusprofundi: Investigating Acetamidase Gene Function
www.nature.com/scientificreports OPEN Developing a genetic manipulation system for the Antarctic archaeon, Halorubrum lacusprofundi: Received: 27 May 2016 Accepted: 16 September 2016 investigating acetamidase gene Published: 06 October 2016 function Y. Liao1, T. J. Williams1, J. C. Walsh2,3, M. Ji1, A. Poljak4, P. M. G. Curmi2, I. G. Duggin3 & R. Cavicchioli1 No systems have been reported for genetic manipulation of cold-adapted Archaea. Halorubrum lacusprofundi is an important member of Deep Lake, Antarctica (~10% of the population), and is amendable to laboratory cultivation. Here we report the development of a shuttle-vector and targeted gene-knockout system for this species. To investigate the function of acetamidase/formamidase genes, a class of genes not experimentally studied in Archaea, the acetamidase gene, amd3, was disrupted. The wild-type grew on acetamide as a sole source of carbon and nitrogen, but the mutant did not. Acetamidase/formamidase genes were found to form three distinct clades within a broad distribution of Archaea and Bacteria. Genes were present within lineages characterized by aerobic growth in low nutrient environments (e.g. haloarchaea, Starkeya) but absent from lineages containing anaerobes or facultative anaerobes (e.g. methanogens, Epsilonproteobacteria) or parasites of animals and plants (e.g. Chlamydiae). While acetamide is not a well characterized natural substrate, the build-up of plastic pollutants in the environment provides a potential source of introduced acetamide. In view of the extent and pattern of distribution of acetamidase/formamidase sequences within Archaea and Bacteria, we speculate that acetamide from plastics may promote the selection of amd/fmd genes in an increasing number of environmental microorganisms. -

Growth of Sedimentary Bathyarchaeota on Lignin As an Energy Source
Growth of sedimentary Bathyarchaeota on lignin as an energy source Tiantian Yua,b,1, Weichao Wuc,d,1, Wenyue Lianga,b, Mark Alexander Levere, Kai-Uwe Hinrichsc,d, and Fengping Wanga,b,2 aState Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, 200240 Shanghai, China; bState Key Laboratory of Ocean Engineering, Shanghai Jiao Tong University, 200240 Shanghai, China; cOrganic Geochemistry Group, MARUM-Center for Marine Environmental Sciences, University of Bremen, 28359 Bremen, Germany; dDepartment of Geosciences, University of Bremen, 28359 Bremen, Germany; and eInstitute of Biogeochemistry and Pollutant Dynamics, Department of Environmental Systems Science, Swiss Federal Institute of Technology Zurich, 8092 Zurich, Switzerland Edited by Edward F. DeLong, University of Hawaii at Manoa, Honolulu, HI, and approved April 16, 2018 (received for review October 30, 2017) Members of the archaeal phylum Bathyarchaeota are among the other hand, acetogenesis from H2/CO2 has been proposed for most abundant microorganisms on Earth. Although versatile met- some lineages of Bathyarchaeota (3). The reductive acetyl-CoA abolic capabilities such as acetogenesis, methanogenesis, and fer- pathway for carbon fixation has been identified in most of the mentation have been suggested for bathyarchaeotal members, no obtained bathyarchaeotal genomes (3, 12, 14). Among these, direct confirmation of these metabolic functions has been achieved members of the bathyarchaeotal subgroups Bathy-3 and Bathy- through growth of Bathyarchaeota in the laboratory. Here we dem- 8 have been suggested to be capable of methanogenesis (12). All onstrate, on the basis of gene-copy numbers and probing of ar- the above studies indicate great versatility in the metabolic po- chaeal lipids, the growth of Bathyarchaeota subgroup Bathy-8 in tentials of Bathyarchaeota, with some lineages possibly being enrichments of estuarine sediments with the biopolymer lignin. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Expression of Cyclomaltodextrinase Gene from Bacillus Halodurans C-125 and Characterization of Its Multisubstrate Specificity
Food Sci. Biotechnol. Vol. 18, No. 3, pp. 776 ~ 781 (2009) ⓒ The Korean Society of Food Science and Technology Expression of Cyclomaltodextrinase Gene from Bacillus halodurans C-125 and Characterization of Its Multisubstrate Specificity Hye-Jeong Kang, Chang-Ku Jeong, Myoung-Uoon Jang, Seung-Ho Choi, Min-Hong Kim1, Jun-Bae Ahn2, Sang-Hwa Lee3, Sook-Ja Jo3, and Tae-Jip Kim* Department of Food Science and Technology, Chungbuk National University, Cheongju, Chungbuk 361-763, Korea 1MH2 Biochemical Co., Ltd., Eumseong, Chungbuk 369-841, Korea 2Department of Food Service Industry, Seowon University, Cheongju, Chungbuk 361-741, Korea 3Department of Food and Nutrition, Seowon University, Cheongju, Chungbuk 361-741, Korea Abstract A putative cyclomaltodextrinase (BHCD) gene was found from the genome of Bacillus halodurans C-125, which encodes 578 amino acids with a predicted molecular mass of 67,279 Da. It shares 42-59% of amino acid sequence identity with common cyclomaltodextrinase (CDase)-family enzymes. The corresponding gene was cloned by polymerase chain reaction (PCR) and the dimeric enzyme with C-terminal 6-histidines was successfully overproduced and purified from recombinant Escherichia coli. BHCD showed the highest activity against β-CD at pH 7.0 and 50oC. Due to its versatile hydrolysis and transglycosylation activities, BHCD has been confirmed as a member of CDases. However, BHCD can be distinguished from other typical CDases on the basis of its novel multisubstrate specificity. While typical CDases have over 10 times higher activity on β-CD than starch or pullulan, the CD-hydrolyzing activity of BHCD is only 2.3 times higher than pullulan. -

Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China
Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China Di Wu Northeast Forestry University Caihong Zhao Northeast Forestry University Hui Bai Forestry Science Research Institute of Heilongjiang Province Fujuan Feng Northeast Forestry University Xin Sui Heilongjiang University Guangyu Sun ( [email protected] ) Northeast Forestry University Research article Keywords: Wetlands, Methanogens, Community diversity, Indicator species, Methanogenic metabolic patterns Posted Date: August 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-54821/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Background: Soil methanogenic microorganisms are one of the primary methane-producing microbes in wetlands. However, we still poorly understand the community characteristic and metabolic patterns of these microorganisms according to vegetation type and seasonal changes. Therefore, to better elucidate the effects of the vegetation type and seasonal factors on the methanogenic community structure and metabolic patterns, we detected the characteristics of the soil methanogenic mcrA gene from three types of natural wetlands in different seasons in the Xiaoxing'an Mountain region, China. Result: The results indicated that the distribution of Methanobacteriaceae (hydrogenotrophic methanogens) was higher in winter, while Methanosarcinaceae and Methanosaetaceae accounted for a higher proportion in summer. Hydrogenotrophic methanogenesis was the dominant trophic pattern in each wetland. The results of principal coordinate analysis and cluster analysis showed that the vegetation type considerably inuenced the methanogenic community composition. The methanogenic community structure in the Betula platyphylla – Larix gmelinii wetland was relatively different from the structure of the other two wetland types. -

Plant Life MagillS Encyclopedia of Science
MAGILLS ENCYCLOPEDIA OF SCIENCE PLANT LIFE MAGILLS ENCYCLOPEDIA OF SCIENCE PLANT LIFE Volume 4 Sustainable Forestry–Zygomycetes Indexes Editor Bryan D. Ness, Ph.D. Pacific Union College, Department of Biology Project Editor Christina J. Moose Salem Press, Inc. Pasadena, California Hackensack, New Jersey Editor in Chief: Dawn P. Dawson Managing Editor: Christina J. Moose Photograph Editor: Philip Bader Manuscript Editor: Elizabeth Ferry Slocum Production Editor: Joyce I. Buchea Assistant Editor: Andrea E. Miller Page Design and Graphics: James Hutson Research Supervisor: Jeffry Jensen Layout: William Zimmerman Acquisitions Editor: Mark Rehn Illustrator: Kimberly L. Dawson Kurnizki Copyright © 2003, by Salem Press, Inc. All rights in this book are reserved. No part of this work may be used or reproduced in any manner what- soever or transmitted in any form or by any means, electronic or mechanical, including photocopy,recording, or any information storage and retrieval system, without written permission from the copyright owner except in the case of brief quotations embodied in critical articles and reviews. For information address the publisher, Salem Press, Inc., P.O. Box 50062, Pasadena, California 91115. Some of the updated and revised essays in this work originally appeared in Magill’s Survey of Science: Life Science (1991), Magill’s Survey of Science: Life Science, Supplement (1998), Natural Resources (1998), Encyclopedia of Genetics (1999), Encyclopedia of Environmental Issues (2000), World Geography (2001), and Earth Science (2001). ∞ The paper used in these volumes conforms to the American National Standard for Permanence of Paper for Printed Library Materials, Z39.48-1992 (R1997). Library of Congress Cataloging-in-Publication Data Magill’s encyclopedia of science : plant life / edited by Bryan D. -

Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma Divulgatum
microorganisms Article Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum Rafael Bargiela 1 , Karin Lanthaler 1,2, Colin M. Potter 1,2 , Manuel Ferrer 3 , Alexander F. Yakunin 1,2, Bela Paizs 1,2, Peter N. Golyshin 1,2 and Olga V. Golyshina 1,2,* 1 School of Natural Sciences, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK; [email protected] (R.B.); [email protected] (K.L.); [email protected] (C.M.P.); [email protected] (A.F.Y.); [email protected] (B.P.); [email protected] (P.N.G.) 2 Centre for Environmental Biotechnology, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK 3 Systems Biotechnology Group, Department of Applied Biocatalysis, CSIC—Institute of Catalysis, Marie Curie 2, 28049 Madrid, Spain; [email protected] * Correspondence: [email protected]; Tel.: +44-1248-388607; Fax: +44-1248-382569 Received: 27 April 2020; Accepted: 15 May 2020; Published: 19 May 2020 Abstract: The archaeon Cuniculiplasma divulgatum is ubiquitous in acidic environments with low-to-moderate temperatures. However, molecular mechanisms underlying its ability to thrive at lower temperatures remain unexplored. Using mass spectrometry (MS)-based proteomics, we analysed the effect of short-term (3 h) exposure to cold. The C. divulgatum genome encodes 2016 protein-coding genes, from which 819 proteins were identified in the cells grown under optimal conditions. In line with the peptidolytic lifestyle of C. divulgatum, its intracellular proteome revealed the abundance of proteases, ABC transporters and cytochrome C oxidase. From 747 quantifiable polypeptides, the levels of 582 proteins showed no change after the cold shock, whereas 104 proteins were upregulated suggesting that they might be contributing to cold adaptation. -

Picrophilus Oshimae and Picrophilus Tomdus Fam. Nov., Gen. Nov., Sp. Nov
INTERNATIONALJOURNAL OF SYSTEMATICBACTERIOLOGY, July 1996, p. 814-816 Vol. 46, No. 3 0020-77 13/96/$04.00+0 Copyright 0 1996, International Union of Microbiological Societies Picrophilus oshimae and Picrophilus tomdus fam. nov., gen. nov., sp. nov., Two Species of Hyperacidophilic, Thermophilic, Heterotrophic, Aerobic Archaea CHRISTA SCHLEPER, GABRIELA PUHLER, HANS- PETER KLENK, AND WOLFRAM ZILLIG* Max Plank Institut fur Biochemie, 0-82152 Martinsried, Germany We describe two species of hyperacidophilic, thermophilic, heterotrophic, aerobic archaea that were isolated from solfataric hydrothermal areas in Hokkaido, Japan. These organisms, Picrophilus oshimae and Picrophilus torridus, represent a novel genus and a novel family, the Picrophilaceae, in the kingdom Euryarchaeota and the order Thermoplasmales. Both of these bacteria are more acidophilic than the genus Thermoplasma since they are able to grow at about pH 0. The moderately thermophilic, hyperacidophilic, aerobic ar- which comprises acid-loving (i.e., hyperacidophilic) organisms. chaea (archaebacteria) (7) Picrophilus oshimae and Picrophilus Separation of these taxa is justified by their phylogenetic dis- rorridus, which have been described previously (4, 5), were tance, (9.5% difference in the 16s rRNA sequences of mem- isolated from moderately hot hydrothermal areas in solfataric bers of the Picrophilaceae and T. acidophilum), by the lack of fields in Hokkaido, Japan. One of the sources of isolation was immunochemical cross-reactions in Ouchterlony immunodif- a solfataric spring which had a temperature of 53°C and a pH fusion assays between the RNA polymerases of P. oshimae and of 2.2, and the other was a rather dry hot soil which had a pH T. acidophilum, which also do not occur between members of of <OS. -

A Korarchaeal Genome Reveals Insights Into the Evolution of the Archaea
A korarchaeal genome reveals insights into the evolution of the Archaea James G. Elkinsa,b, Mircea Podarc, David E. Grahamd, Kira S. Makarovae, Yuri Wolfe, Lennart Randauf, Brian P. Hedlundg, Ce´ line Brochier-Armaneth, Victor Kunini, Iain Andersoni, Alla Lapidusi, Eugene Goltsmani, Kerrie Barryi, Eugene V. Koonine, Phil Hugenholtzi, Nikos Kyrpidesi, Gerhard Wannerj, Paul Richardsoni, Martin Kellerc, and Karl O. Stettera,k,l aLehrstuhl fu¨r Mikrobiologie und Archaeenzentrum, Universita¨t Regensburg, D-93053 Regensburg, Germany; cBiosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831; dDepartment of Chemistry and Biochemistry, University of Texas, Austin, TX 78712; eNational Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894; fDepartment of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520; gSchool of Life Sciences, University of Nevada, Las Vegas, NV 89154; hLaboratoire de Chimie Bacte´rienne, Unite´ Propre de Recherche 9043, Centre National de la Recherche Scientifique, Universite´de Provence Aix-Marseille I, 13331 Marseille Cedex 3, France; iU.S. Department of Energy Joint Genome Institute, Walnut Creek, CA 94598; jInstitute of Botany, Ludwig Maximilians University of Munich, D-80638 Munich, Germany; and kInstitute of Geophysics and Planetary Physics, University of California, Los Angeles, CA 90095 Communicated by Carl R. Woese, University of Illinois at Urbana–Champaign, Urbana, IL, April 2, 2008 (received for review January 7, 2008) The candidate division Korarchaeota comprises a group of uncul- and sediment samples from Obsidian Pool as an inoculum. The tivated microorganisms that, by their small subunit rRNA phylog- cultivation system supported the stable growth of a mixed commu- eny, may have diverged early from the major archaeal phyla nity of hyperthermophilic bacteria and archaea including an or- Crenarchaeota and Euryarchaeota. -
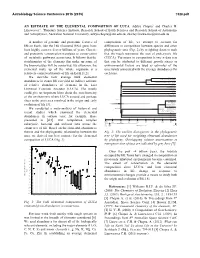
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Archaeal Adaptation to Higher Temperatures Revealed by Genomic Sequence of Thermoplasma Volcanium
Archaeal adaptation to higher temperatures revealed by genomic sequence of Thermoplasma volcanium Tsuyoshi Kawashima*†, Naoki Amano*†‡, Hideaki Koike*†, Shin-ichi Makino†, Sadaharu Higuchi†, Yoshie Kawashima-Ohya†, Koji Watanabe§, Masaaki Yamazaki§, Keiichi Kanehori¶, Takeshi Kawamotoʈ, Tatsuo Nunoshiba**, Yoshihiro Yamamoto††, Hironori Aramaki‡‡, Kozo Makino§§, and Masashi Suzuki†¶¶ †National Institute of Bioscience and Human Technology, Core Research for Evolutional Science and Technology Centre of Structural Biology, 1-1 Higashi, Tsukuba 305-0046, Japan; ‡Doctoral Program in Medical Sciences, University of Tsukuba, 1-1-1 Tennohdai, Tsukuba 305-0006, Japan; §Bioscience Research Laboratory, Fujiya, 228 Soya, Hadano 257-0031, Japan; ¶DNA Analysis Department, Techno Research Laboratory, Hitachi Science Systems, 1-280 Higashi-Koigakubo, Kokubunji 185-8601, Japan; ʈDepartment of Biochemistry, Hiroshima University, School of Dentistry, 1-2-3 Kasumi, Minami-ku, Hiroshima 734-8553, Japan; **Department of Molecular and Cellular Biology, Biological Institute, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan; ††Department of Genetics, Hyogo College of Medicine, Nishinomiya 663-8501, Japan; ‡‡Department of Molecular Biology, Daiichi College of Pharmaceutical Science, 22-1 Tamagawa-cho, Minami-ku, Fukuoka 815-8511, Japan; and §§Department of Molecular Microbiology, The Research Institute of Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita 565-0871, Japan Edited by Aaron Klug, Royal Society of London, London, United Kingdom, and approved October 16, 2000 (received for review August 18, 2000) The complete genomic sequence of the archaeon Thermoplasma contigs. The remaining gaps were bridged by DNA fragments volcanium, possessing optimum growth temperature (OGT) of constructed using the PCR. The average repetition in sequencing 60°C, is reported. By systematically comparing this genomic se- the same base positions was 13-fold. -

The Main (Glyco) Phospholipid (MPL) of Thermoplasma Acidophilum
International Journal of Molecular Sciences Review The Main (Glyco) Phospholipid (MPL) of Thermoplasma acidophilum Hans-Joachim Freisleben 1,2 1 Goethe-Universität, Gustav-Embden-Zentrum, Laboratory of Microbiological Chemistry, Theodor-Stern-Kai 7, D-60590 Frankfurt am Main, Germany; [email protected] 2 Universitas Indonesia, Medical Research Unit, Faculty of Medicine, Jalan Salemba Raya 6, Jakarta 10430, Indonesia Received: 19 September 2019; Accepted: 18 October 2019; Published: 21 October 2019 Abstract: The main phospholipid (MPL) of Thermoplasma acidophilum DSM 1728 was isolated, purified and physico-chemically characterized by differential scanning calorimetry (DSC)/differential thermal analysis (DTA) for its thermotropic behavior, alone and in mixtures with other lipids, cholesterol, hydrophobic peptides and pore-forming ionophores. Model membranes from MPL were investigated; black lipid membrane, Langmuir-Blodgett monolayer, and liposomes. Laboratory results were compared to computer simulation. MPL forms stable and resistant liposomes with highly proton-impermeable membrane and mixes at certain degree with common bilayer-forming lipids. Monomeric bacteriorhodopsin and ATP synthase from Micrococcus luteus were co-reconstituted and light-driven ATP synthesis measured. This review reports about almost four decades of research on Thermoplasma membrane and its MPL as well as transfer of this research to Thermoplasma species recently isolated from Indonesian volcanoes. Keywords: Thermoplasma acidophilum; Thermoplasma volcanium;