Protein Phosphatases Targeted Library
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ATP-Induced Focal Adhesion Kinase Activity Is Negatively Modulated by Phospholipase D2 in PC12 Cells
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 33, No. 3, 150-155, September 2001 ATP-induced focal adhesion kinase activity is negatively modulated by phospholipase D2 in PC12 cells Yoe-Sik Bae1 and Sung Ho Ryu1,2 Introduction 1 Division of Molecular and Life Sciences, Pohang University of Purinergic receptors have been reported to play impor- Science and Technology, Pohang 790-784, Korea tant roles on the regulation of neuronal cell functions 2 Corresponding author: Tel, +82-54-279-2292; (Communi et al., 2000; Di Iorio et al., 1998). ATP, a Fax, +82-54-279-2199; E-mail, [email protected] ligand for the receptors modulate various cellular re- sponses such as mitogenic and morphogenic activity in Accepted 18 September 2001 PC12 rat pheochromocytoma cells (Neary et al., 1996; Soltoff et al., 1998; Schindelholz et al., 2000). Stimu- Abbreviations: Fak, focal adhesion kinase; PLD, phospholipase D; lation of cells with ATP induces tyrosine phosphorylation PA, phosphatidic acid; PC, phosphatidylcholine; DAG, diacylglyc- of several cytoskeletal proteins and focal adhesion erol; PBt, phosphatidylbutanol; PKC, protein kinase C; PAP, phos- molecules such as focal adhesion kinase (Fak), proline- phatidic acid phosphohydrolase rich tyrosine kinase (Pyk2), and paxillin (Soltoff et al., 1998; Schindelholz et al., 2000). Since these cytosk- eleton-associated proteins have been regarded as important factors for the regulation of neuronal cell Abstract functions, the study on the regulatory mechanism for the proteins remains an important issue. Extracellular ATP has been known to modulate vari- Phospholipase D (PLD) catalyzes the hydrolysis of ous cellular responses including mitogenesis, secre- phosphatidylcholine (PC) into phosphatidic acid (PA) tion and morphogenic activity in neuronal cells. -

Datasheet for Protein Phosphatase 1 (PP1) (P0754; Lot 0121306)
Supplied in: 200 mM NaCl, 50 mM HEPES Unit Definition: One unit is defined as Notes On Use: Avoid freeze/thaw cycles. Can be Protein the amount of enzyme that hydrolyzes 1 nmol of stored for 1 week or less at –20°C. (pH 7.0 @ 25°C), 1 mM MnCl2, 0.1 mM EGTA, Phosphatase 1 2.5 mM dithiothreitol, 0.025% Tween-20 and p-Nitrophenyl Phosphate (50 mM) (NEB #P0757) 50% glycerol. Store at –70°C in 1 minute at 30°C in a total reaction volume of The following information can be used as (PP1) 50 µl. suggested initial conditions for dephosphorylation 1-800-632-7799 Applications: PP1 can be used to release of proteins with PP1. [email protected] phosphate groups from phosphorylated serine, Specific Activity: ~ 80,000 units/mg. www.neb.com 0.1 unit of PP1 removes ~100% of phosphates P0754S 012130614061 threonine and tyrosine residues in proteins. Note that different proteins are dephosphorylated at Molecular Weight: 37.5 kDa. (0.5 nmol) from phosphoserine/threonine different rates. residues in phosphorylase a as well as in P0754S r y Purity: PP1 has been purified to > 90% phosphorylated myelin basic protein (phospho- 100 units 2,500 U/ml Lot: 0121306 Reagents Supplied with Enzyme: homogeneity as determined by SDS-PAGE and MyBP, 18.5 kDa) in 30 minutes in a 50 µl reaction. RECOMBINANT Store at –70°C Exp: 6/14 10X NEBuffer for Protein MetalloPhosphatases Coomassie Blue staining. The concentration of phospho-MyBP is 10 µM (PMP) with respect to phosphate. Description: Protein Phosphatase 1 (PP1) is a 10X MnCl2 (10 mM) Quality Assurance: PP1 contains no detectable Mn2+-dependent protein phosphatase with activity protease activity. -

Role of Phospholipases in Adrenal Steroidogenesis
229 1 W B BOLLAG Phospholipases in adrenal 229:1 R29–R41 Review steroidogenesis Role of phospholipases in adrenal steroidogenesis Wendy B Bollag Correspondence should be addressed Charlie Norwood VA Medical Center, One Freedom Way, Augusta, GA, USA to W B Bollag Department of Physiology, Medical College of Georgia, Augusta University (formerly Georgia Regents Email University), Augusta, GA, USA [email protected] Abstract Phospholipases are lipid-metabolizing enzymes that hydrolyze phospholipids. In some Key Words cases, their activity results in remodeling of lipids and/or allows the synthesis of other f adrenal cortex lipids. In other cases, however, and of interest to the topic of adrenal steroidogenesis, f angiotensin phospholipases produce second messengers that modify the function of a cell. In this f intracellular signaling review, the enzymatic reactions, products, and effectors of three phospholipases, f phospholipids phospholipase C, phospholipase D, and phospholipase A2, are discussed. Although f signal transduction much data have been obtained concerning the role of phospholipases C and D in regulating adrenal steroid hormone production, there are still many gaps in our knowledge. Furthermore, little is known about the involvement of phospholipase A2, Endocrinology perhaps, in part, because this enzyme comprises a large family of related enzymes of that are differentially regulated and with different functions. This review presents the evidence supporting the role of each of these phospholipases in steroidogenesis in the Journal Journal of Endocrinology adrenal cortex. (2016) 229, R1–R13 Introduction associated GTP-binding protein exchanges a bound GDP for a GTP. The G protein with GTP bound can then Phospholipids serve a structural function in the cell in that activate the enzyme, phospholipase C (PLC), that cleaves they form the lipid bilayer that maintains cell integrity. -

Kinome Profiling of Clinical Cancer Specimens
Published OnlineFirst March 23, 2010; DOI: 10.1158/0008-5472.CAN-09-3989 Review Cancer Research Kinome Profiling of Clinical Cancer Specimens Kaushal Parikh and Maikel P. Peppelenbosch Abstract Over the past years novel technologies have emerged to enable the determination of the transcriptome and proteome of clinical samples. These data sets will prove to be of significant value to our elucidation of the mechanisms that govern pathophysiology and may provide biological markers for future guidance in person- alized medicine. However, an equally important goal is to define those proteins that participate in signaling pathways during the disease manifestation itself or those pathways that are made active during successful clinical treatment of the disease: the main challenge now is the generation of large-scale data sets that will allow us to define kinome profiles with predictive properties on the outcome-of-disease and to obtain insight into tissue-specific analysis of kinase activity. This review describes the current techniques available to gen- erate kinome profiles of clinical tissue samples and discusses the future strategies necessary to achieve new insights into disease mechanisms and treatment targets. Cancer Res; 70(7); 2575–8. ©2010 AACR. Background the current state of the cell or tissue as characterized by pro- teomic and metabolic measurements (3). The past five years have seen an exponential increase in In this review we describe and evaluate the various tech- technological development to explore the genome and the niques that have recently become available to study the ki- proteome to understand the molecular basis of disease. How- nomes of clinical tissue samples. -
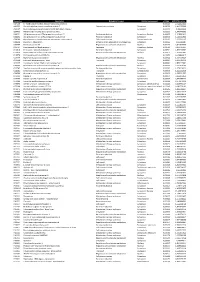
Accession Description Biological Process Cellular Component P-Value SCZ/CTRL Ration A4FU69 EF-Hand Calcium-Binding Domain-Contai
Accession Description Biological Process Cellular component p-Value SCZ/CTRL ration A4FU69 EF-hand calcium-binding domain-containing protein 5 0,001101 2,724427411 A4UGR9 Xin actin-binding repeat-containing protein 2 Cytoskeletal anchoring Cytoplasm 0,006756 1,413953388 A6NCE7 Microtubule-associated proteins 1A/1B light chain 3 beta 2 Cytoplasm 0,001417 1,99612969 B2RPK0 Putative high mobility group protein B1-like 1 0,032314 1,590743352 O00231 26S proteasome non-ATPase regulatory subunit 11 Protein metabolism Cytoplasm; Nucleus 0,000029 1,428664042 O00232 26S proteasome non-ATPase regulatory subunit 12 Protein metabolism Cytoplasm 0,008566 1,544545922 O00264 Membrane-associated progesterone receptor component 1 Cell communication Plasma membrane 0,001459 2,322924147 O00429 Dynamin-1-like protein Mitochondrion organization and biogenesis Cytoplasm 0,006560 0,06391487 O00567 Nucleolar protein 56 Regulation of nucleotide metabolism Nucleus 0,007330 3,842896338 O14737 Programmed cell death protein 5 Apoptosis Cytoplasm; Nucleus 0,006358 3,836727237 O14818 Proteasome subunit alpha type-7 Protein metabolism Cytoplasm 0,030521 1,893928387 O14979 Heterogeneous nuclear ribonucleoprotein D-like Regulation of nucleotide metabolism Nucleus 0,000637 2,150005885 O15078 Centrosomal protein of 290 kDa 0,015359 14,26648619 O15347 High mobility group protein B3 Regulation of nucleotide metabolism Nucleus 0,005500 1,364309014 O15540 Fatty acid-binding protein_ brain Transport Cytoplasm 0,000087 3,125786118 O43237 Cytoplasmic dynein 1 light intermediate -

Protein Kinases Phosphorylation/Dephosphorylation Protein Phosphorylation Is One of the Most Important Mechanisms of Cellular Re
Protein Kinases Phosphorylation/dephosphorylation Protein phosphorylation is one of the most important mechanisms of cellular responses to growth, stress metabolic and hormonal environmental changes. Most mammalian protein kinases have highly a homologous 30 to 32 kDa catalytic domain. • Most common method of reversible modification - activation and localization • Up to 1/3 of cellular proteins can be phosphorylated • Leads to a very fast response to cellular stress, hormonal changes, learning processes, transcription regulation .... • Different than allosteric or Michealis Menten regulation Protein Kinome To date – 518 human kinases known • 50 kinase families between yeast, invertebrate and mammaliane kinomes • 518 human PKs, most (478) belong to single super family whose catalytic domain are homologous. • Kinase dendrogram displays relative similarities based on catalytic domains. • AGC (PKA, PKG, PKC) • CAMK (Casein kinase 1) • CMGC (CDC, MAPK, GSK3, CLK) • STE (Sterile 7, 11 & 20 kinases) • TK (Tryosine kinases memb and cyto) • TKL (Tyrosine kinase-like) • Phosphorylation stabilized thermodynamically - only half available energy used in adding phosphoryl to protein - change in free energy forces phosphorylation reaction in one direction • Phosphatases reverse direction • The rate of reaction of most phosphatases are 1000 times faster • Phosphorylation occurs on Ser/The or Tyr • What differences occur due to the addition of a phosphoryl group? • Regulation of protein phosphorylation varies depending on protein - some turned on or off -

The Regulatory Roles of Phosphatases in Cancer
Oncogene (2014) 33, 939–953 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW The regulatory roles of phosphatases in cancer J Stebbing1, LC Lit1, H Zhang, RS Darrington, O Melaiu, B Rudraraju and G Giamas The relevance of potentially reversible post-translational modifications required for controlling cellular processes in cancer is one of the most thriving arenas of cellular and molecular biology. Any alteration in the balanced equilibrium between kinases and phosphatases may result in development and progression of various diseases, including different types of cancer, though phosphatases are relatively under-studied. Loss of phosphatases such as PTEN (phosphatase and tensin homologue deleted on chromosome 10), a known tumour suppressor, across tumour types lends credence to the development of phosphatidylinositol 3--kinase inhibitors alongside the use of phosphatase expression as a biomarker, though phase 3 trial data are lacking. In this review, we give an updated report on phosphatase dysregulation linked to organ-specific malignancies. Oncogene (2014) 33, 939–953; doi:10.1038/onc.2013.80; published online 18 March 2013 Keywords: cancer; phosphatases; solid tumours GASTROINTESTINAL MALIGNANCIES abs in sera were significantly associated with poor survival in Oesophageal cancer advanced ESCC, suggesting that they may have a clinical utility in Loss of PTEN (phosphatase and tensin homologue deleted on ESCC screening and diagnosis.5 chromosome 10) expression in oesophageal cancer is frequent, Cao et al.6 investigated the role of protein tyrosine phosphatase, among other gene alterations characterizing this disease. Zhou non-receptor type 12 (PTPN12) in ESCC and showed that PTPN12 et al.1 found that overexpression of PTEN suppresses growth and protein expression is higher in normal para-cancerous tissues than induces apoptosis in oesophageal cancer cell lines, through in 20 ESCC tissues. -

The Murine Orthologue of the Golgi-Localized TPTE Protein Provides Clues to the Evolutionary History of the Human TPTE Gene Family
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Hum Genet (2001) 109:569–575 DOI 10.1007/s004390100607 ORIGINAL INVESTIGATION Michel Guipponi · Caroline Tapparel · Olivier Jousson · Nathalie Scamuffa · Christophe Mas · Colette Rossier · Pierre Hutter · Paolo Meda · Robert Lyle · Alexandre Reymond · Stylianos E. Antonarakis The murine orthologue of the Golgi-localized TPTE protein provides clues to the evolutionary history of the human TPTE gene family Received: 5 June 2001 / Accepted: 17 August 2001 / Published online: 27 October 2001 © Springer-Verlag 2001 Abstract The human TPTE gene encodes a testis-spe- events. The Y chromosome copy of TPTE is a pseudo- cific protein that contains four potential transmembrane gene and is not therefore involved in the testis expression domains and a protein tyrosine phosphatase motif, and of this gene family. shows homology to the tumor suppressor PTEN/MMAC1. Chromosomal mapping revealed multiple copies of the TPTE gene present on the acrocentric chromosomes 13, Introduction 15, 21 and 22, and the Y chromosome. Zooblot analysis suggests that mice may possess only one copy of TPTE. We have recently identified a testis-specific cDNA, TPTE In the present study, we report the isolation and initial (Transmembrane Phosphatase with TEnsin homology), characterization of the full-length cDNA of the mouse ho- that encodes a predicted protein of 551 amino acids con- mologue Tpte. At least three different mRNA transcripts taining four potential transmembrane domains and a tyro- (Tpte.a, b, c) are produced via alternative splicing, encod- sine phosphatase motif (Chen H et al. -

Inhibition of Protein Phosphatase 1 Translation by Preventing JNK
Active ERK Contributes to Protein Translation by Preventing JNK-Dependent Inhibition of Protein Phosphatase 1 This information is current as Martha M. Monick, Linda S. Powers, Thomas J. Gross, of September 28, 2021. Dawn M. Flaherty, Christopher W. Barrett and Gary W. Hunninghake J Immunol 2006; 177:1636-1645; ; doi: 10.4049/jimmunol.177.3.1636 http://www.jimmunol.org/content/177/3/1636 Downloaded from References This article cites 59 articles, 43 of which you can access for free at: http://www.jimmunol.org/content/177/3/1636.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Active ERK Contributes to Protein Translation by Preventing JNK-Dependent Inhibition of Protein Phosphatase 11 Martha M. Monick,2 Linda S. Powers, Thomas J. Gross, Dawn M. Flaherty, Christopher W. -

Identification of Protein O-Glcnacylation Sites Using Electron Transfer Dissociation Mass Spectrometry on Native Peptides
Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides Robert J. Chalkleya, Agnes Thalhammerb, Ralf Schoepfera,b, and A. L. Burlingamea,1 aDepartment of Pharmaceutical Chemistry, University of California, 600 16th Street, Genentech Hall, Suite N472, San Francisco, CA 94158; and bLaboratory for Molecular Pharmacology, and Department of Neuroscience, Physiology, and Pharmacology, University College London, Gower Street, London WC1E 6BT, United Kingdom Edited by James A. Wells, University of California, San Francisco, CA, and approved April 15, 2009 (received for review January 12, 2009) Protein O-GlcNAcylation occurs in all animals and plants and is and synaptic plasticity have been found O-GlcNAcylated (8). The implicated in modulation of a wide range of cytosolic and nuclear importance of O-GlcNAcylation is highlighted by the brain-specific protein functions, including gene silencing, nutrient and stress sens- OGT knockout mouse model that displays developmental defects ing, phosphorylation signaling, and diseases such as diabetes and that result in neonatal death (9). Most interestingly, in analogy to Alzheimer’s. The limiting factor impeding rapid progress in decipher- phosphorylation, synaptic activity leads to a dynamic modulation of ing the biological functions of protein O-GlcNAcylation has been the O-GlcNAc levels (10) and is also involved in long-term potentiation inability to easily identify exact residues of modification. We describe and memory (11). a robust, high-sensitivity strategy able to assign O-GlcNAcylation sites Phosphorylation is the most heavily studied regulatory PTM, and of native modified peptides using electron transfer dissociation mass proteomic approaches for global enrichment, detection, and char- spectrometry. -

Genetic Alterations of Protein Tyrosine Phosphatases in Human Cancers
Oncogene (2015) 34, 3885–3894 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc REVIEW Genetic alterations of protein tyrosine phosphatases in human cancers S Zhao1,2,3, D Sedwick3,4 and Z Wang2,3 Protein tyrosine phosphatases (PTPs) are enzymes that remove phosphate from tyrosine residues in proteins. Recent whole-exome sequencing of human cancer genomes reveals that many PTPs are frequently mutated in a variety of cancers. Among these mutated PTPs, PTP receptor T (PTPRT) appears to be the most frequently mutated PTP in human cancers. Beside PTPN11, which functions as an oncogene in leukemia, genetic and functional studies indicate that most of mutant PTPs are tumor suppressor genes. Identification of the substrates and corresponding kinases of the mutant PTPs may provide novel therapeutic targets for cancers harboring these mutant PTPs. Oncogene (2015) 34, 3885–3894; doi:10.1038/onc.2014.326; published online 29 September 2014 INTRODUCTION tyrosine/threonine-specific phosphatases. (4) Class IV PTPs include Protein tyrosine phosphorylation has a critical role in virtually all four Drosophila Eya homologs (Eya1, Eya2, Eya3 and Eya4), which human cellular processes that are involved in oncogenesis.1 can dephosphorylate both tyrosine and serine residues. Protein tyrosine phosphorylation is coordinately regulated by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases 1 THE THREE-DIMENSIONAL STRUCTURE AND CATALYTIC (PTPs). Although PTKs add phosphate to tyrosine residues in MECHANISM OF PTPS proteins, PTPs remove it. Many PTKs are well-documented oncogenes.1 Recent cancer genomic studies provided compelling The three-dimensional structures of the catalytic domains of evidence that many PTPs function as tumor suppressor genes, classical PTPs (RPTPs and non-RPTPs) are extremely well because a majority of PTP mutations that have been identified in conserved.5 Even the catalytic domain structures of the dual- human cancers are loss-of-function mutations. -

GSK3 and Its Interactions with the PI3K/AKT/Mtor Signalling Network
Heriot-Watt University Research Gateway GSK3 and its interactions with the PI3K/AKT/mTOR signalling network Citation for published version: Hermida, MA, Kumar, JD & Leslie, NR 2017, 'GSK3 and its interactions with the PI3K/AKT/mTOR signalling network', Advances in Biological Regulation, vol. 65, pp. 5-15. https://doi.org/10.1016/j.jbior.2017.06.003 Digital Object Identifier (DOI): 10.1016/j.jbior.2017.06.003 Link: Link to publication record in Heriot-Watt Research Portal Document Version: Peer reviewed version Published In: Advances in Biological Regulation Publisher Rights Statement: © 2017 Elsevier B.V. General rights Copyright for the publications made accessible via Heriot-Watt Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy Heriot-Watt University has made every reasonable effort to ensure that the content in Heriot-Watt Research Portal complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 27. Sep. 2021 Accepted Manuscript GSK3 and its interactions with the PI3K/AKT/mTOR signalling network Miguel A. Hermida, J. Dinesh Kumar, Nick R. Leslie PII: S2212-4926(17)30124-0 DOI: 10.1016/j.jbior.2017.06.003 Reference: JBIOR 180 To appear in: Advances in Biological Regulation Received Date: 13 June 2017 Accepted Date: 23 June 2017 Please cite this article as: Hermida MA, Dinesh Kumar J, Leslie NR, GSK3 and its interactions with the PI3K/AKT/mTOR signalling network, Advances in Biological Regulation (2017), doi: 10.1016/ j.jbior.2017.06.003.