Adeno-Associated Virus Rep Proteins Antagonize Phosphatase PP1 To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ATP-Induced Focal Adhesion Kinase Activity Is Negatively Modulated by Phospholipase D2 in PC12 Cells
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 33, No. 3, 150-155, September 2001 ATP-induced focal adhesion kinase activity is negatively modulated by phospholipase D2 in PC12 cells Yoe-Sik Bae1 and Sung Ho Ryu1,2 Introduction 1 Division of Molecular and Life Sciences, Pohang University of Purinergic receptors have been reported to play impor- Science and Technology, Pohang 790-784, Korea tant roles on the regulation of neuronal cell functions 2 Corresponding author: Tel, +82-54-279-2292; (Communi et al., 2000; Di Iorio et al., 1998). ATP, a Fax, +82-54-279-2199; E-mail, [email protected] ligand for the receptors modulate various cellular re- sponses such as mitogenic and morphogenic activity in Accepted 18 September 2001 PC12 rat pheochromocytoma cells (Neary et al., 1996; Soltoff et al., 1998; Schindelholz et al., 2000). Stimu- Abbreviations: Fak, focal adhesion kinase; PLD, phospholipase D; lation of cells with ATP induces tyrosine phosphorylation PA, phosphatidic acid; PC, phosphatidylcholine; DAG, diacylglyc- of several cytoskeletal proteins and focal adhesion erol; PBt, phosphatidylbutanol; PKC, protein kinase C; PAP, phos- molecules such as focal adhesion kinase (Fak), proline- phatidic acid phosphohydrolase rich tyrosine kinase (Pyk2), and paxillin (Soltoff et al., 1998; Schindelholz et al., 2000). Since these cytosk- eleton-associated proteins have been regarded as important factors for the regulation of neuronal cell Abstract functions, the study on the regulatory mechanism for the proteins remains an important issue. Extracellular ATP has been known to modulate vari- Phospholipase D (PLD) catalyzes the hydrolysis of ous cellular responses including mitogenesis, secre- phosphatidylcholine (PC) into phosphatidic acid (PA) tion and morphogenic activity in neuronal cells. -

Datasheet for Protein Phosphatase 1 (PP1) (P0754; Lot 0121306)
Supplied in: 200 mM NaCl, 50 mM HEPES Unit Definition: One unit is defined as Notes On Use: Avoid freeze/thaw cycles. Can be Protein the amount of enzyme that hydrolyzes 1 nmol of stored for 1 week or less at –20°C. (pH 7.0 @ 25°C), 1 mM MnCl2, 0.1 mM EGTA, Phosphatase 1 2.5 mM dithiothreitol, 0.025% Tween-20 and p-Nitrophenyl Phosphate (50 mM) (NEB #P0757) 50% glycerol. Store at –70°C in 1 minute at 30°C in a total reaction volume of The following information can be used as (PP1) 50 µl. suggested initial conditions for dephosphorylation 1-800-632-7799 Applications: PP1 can be used to release of proteins with PP1. [email protected] phosphate groups from phosphorylated serine, Specific Activity: ~ 80,000 units/mg. www.neb.com 0.1 unit of PP1 removes ~100% of phosphates P0754S 012130614061 threonine and tyrosine residues in proteins. Note that different proteins are dephosphorylated at Molecular Weight: 37.5 kDa. (0.5 nmol) from phosphoserine/threonine different rates. residues in phosphorylase a as well as in P0754S r y Purity: PP1 has been purified to > 90% phosphorylated myelin basic protein (phospho- 100 units 2,500 U/ml Lot: 0121306 Reagents Supplied with Enzyme: homogeneity as determined by SDS-PAGE and MyBP, 18.5 kDa) in 30 minutes in a 50 µl reaction. RECOMBINANT Store at –70°C Exp: 6/14 10X NEBuffer for Protein MetalloPhosphatases Coomassie Blue staining. The concentration of phospho-MyBP is 10 µM (PMP) with respect to phosphate. Description: Protein Phosphatase 1 (PP1) is a 10X MnCl2 (10 mM) Quality Assurance: PP1 contains no detectable Mn2+-dependent protein phosphatase with activity protease activity. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Brd4 Bridges the Transcriptional Regulators, Aire and P-Tefb, to Promote Elongation of Peripheral-Tissue Antigen Transcripts In
Brd4 bridges the transcriptional regulators, Aire and PNAS PLUS P-TEFb, to promote elongation of peripheral-tissue antigen transcripts in thymic stromal cells Hideyuki Yoshidaa, Kushagra Bansala, Uwe Schaeferb, Trevor Chapmanc, Inmaculada Riojac, Irina Proektd, Mark S. Andersone, Rab K. Prinjhac, Alexander Tarakhovskyb, Christophe Benoista,f,1, and Diane Mathisa,f,1 aDivision of Immunology, Department of Microbiology and Immunobiology, Harvard Medical School, Boston, MA 02115; bLaboratory of Immune Cell Epigenetics and Signaling, The Rockefeller University, New York, NY 10065; cEpinova Discovery Performance Unit, Immuno-Inflammation Therapy Area, Medicines Research Centre, GlaxoSmithKline, Stevenage SG1 2NY, United Kingdom; dDepartment of Microbiology and Immunology, University of California, San Francisco, CA 94143; eDiabetes Center, University of California, San Francisco, CA 94143; and fEvergrande Center for Immunologic Diseases, Harvard Medical School and Brigham and Women’s Hospital, Boston, MA 02115 Contributed by Diane Mathis, June 22, 2015 (sent for review April 20, 2015; reviewed by Leslie J. Berg and Tadatsugu Taniguchi) Aire controls immunologic tolerance by inducing a battery of turning transcription up or down. Indeed, recent data derived thymic transcripts encoding proteins characteristic of peripheral from a variety of experimental approaches argue that Aire’smajor tissues. Its unusually broad effect is achieved by releasing RNA modus operandi is to release promoter-proximal RNA polymerase II polymerase II paused just downstream of transcriptional start (Pol-II) pausing (11–13). sites. We explored Aire’s collaboration with the bromodomain- It has become increasingly clear that the regulation of Pol-II containing protein, Brd4, uncovering an astonishing correspon- pausing is a major nexus of transcriptional control (14), and the dencebetweenthosegenesinducedbyAireandthoseinhibited focus of transcription factors as diverse as NF-κB (15), c-Myc (16), by a small-molecule bromodomain blocker. -

Structure of the Tripartite Motif of KAP1/TRIM28 Identifies Molecular Interfaces Required for Transcriptional Silencing of Retrotransposons
bioRxiv preprint doi: https://doi.org/10.1101/505677; this version posted December 25, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Structure of the tripartite motif of KAP1/TRIM28 identifies molecular interfaces required for transcriptional silencing of retrotransposons Guido A. Stoll1, Shun-ichiro Oda1, Zheng-Shan Chong1, Minmin Yu2, Stephen H. McLaughlin2 and Yorgo Modis1,* 1 Molecular Immunity Unit, Department of Medicine, University of Cambridge, MRC Laboratory of Molecular Biology, Cambridge Biomedical Campus, Cambridge, CB2 0QH, UK 2 MRC Laboratory oF Molecular Biology, Cambridge Biomedical Campus, Cambridge, CB2 0QH, UK * Correspondence: [email protected] (Y.M.) Abstract Transcription oF transposable elements is tightly regulated to prevent damage to the genome. The family of KRAB domain-containing zinc Finger proteins (KRAB-ZFPs) and KRAB-associated protein 1 (KAP1/TRIM28) play a key role in regulating retrotransposons. KRAB-ZFPs recognize speciFic retrotransposon sequences and recruit KAP1, which controls the assembly of an epigenetic silencing complex including histone H3K9 methyltransferase SETDB1. The chromatin remodeling activities of this complex repress transcription of the targeted transposable element and any adjacent genes. Here, we use biophysical and structural approaches to show that the tripartite motif (TRIM) of KAP1 Forms antiparallel dimers, which Further assemble into tetramers and higher-order oligomers in a concentration-dependent manner. Structure-based mutations in the B-box 1 domain prevented higher-order oligomerization without a signiFicant loss oF retrotransposon silencing activity in a cell-based assay, indicating that, in contrast to other TRIM family members, selF-assembly is not essential For the function of KAP1. -

TRIM28 Is Required by the Mouse KRAB Domain Protein ZFP568 to Control Convergent Extension and Morphogenesis of Extra-Embryonic Tissues Maho Shibata1, Kristin E
RESEARCH ARTICLE 5333 Development 138, 5333-5343 (2011) doi:10.1242/dev.072546 © 2011. Published by The Company of Biologists Ltd TRIM28 is required by the mouse KRAB domain protein ZFP568 to control convergent extension and morphogenesis of extra-embryonic tissues Maho Shibata1, Kristin E. Blauvelt1, Karel F. Liem, Jr2 and María J. García-García1,* SUMMARY TRIM28 is a transcriptional regulator that is essential for embryonic development and is implicated in a variety of human diseases. The roles of TRIM28 in distinct biological processes are thought to depend on its interaction with factors that determine its DNA target specificity. However, functional evidence linking TRIM28 to specific co-factors is scarce. chatwo, a hypomorphic allele of Trim28, causes embryonic lethality and defects in convergent extension and morphogenesis of extra-embryonic tissues. These phenotypes are remarkably similar to those of mutants in the Krüppel-associated box (KRAB) zinc finger protein ZFP568, providing strong genetic evidence that ZFP568 and TRIM28 control morphogenesis through a common molecular mechanism. We determined that chatwo mutations decrease TRIM28 protein stability and repressive activity, disrupting both ZFP568-dependent and ZFP568-independent roles of TRIM28. These results, together with the analysis of embryos bearing a conditional inactivation of Trim28 in embryonic-derived tissues, revealed that TRIM28 is differentially required by ZFP568 and other factors during the early stages of mouse embryogenesis. In addition to uncovering novel roles of TRIM28 in convergent extension and morphogenesis of extra-embryonic tissues, our characterization of chatwo mutants demonstrates that KRAB domain proteins are essential to determine some of the biological functions of TRIM28. -

Role of Phospholipases in Adrenal Steroidogenesis
229 1 W B BOLLAG Phospholipases in adrenal 229:1 R29–R41 Review steroidogenesis Role of phospholipases in adrenal steroidogenesis Wendy B Bollag Correspondence should be addressed Charlie Norwood VA Medical Center, One Freedom Way, Augusta, GA, USA to W B Bollag Department of Physiology, Medical College of Georgia, Augusta University (formerly Georgia Regents Email University), Augusta, GA, USA [email protected] Abstract Phospholipases are lipid-metabolizing enzymes that hydrolyze phospholipids. In some Key Words cases, their activity results in remodeling of lipids and/or allows the synthesis of other f adrenal cortex lipids. In other cases, however, and of interest to the topic of adrenal steroidogenesis, f angiotensin phospholipases produce second messengers that modify the function of a cell. In this f intracellular signaling review, the enzymatic reactions, products, and effectors of three phospholipases, f phospholipids phospholipase C, phospholipase D, and phospholipase A2, are discussed. Although f signal transduction much data have been obtained concerning the role of phospholipases C and D in regulating adrenal steroid hormone production, there are still many gaps in our knowledge. Furthermore, little is known about the involvement of phospholipase A2, Endocrinology perhaps, in part, because this enzyme comprises a large family of related enzymes of that are differentially regulated and with different functions. This review presents the evidence supporting the role of each of these phospholipases in steroidogenesis in the Journal Journal of Endocrinology adrenal cortex. (2016) 229, R1–R13 Introduction associated GTP-binding protein exchanges a bound GDP for a GTP. The G protein with GTP bound can then Phospholipids serve a structural function in the cell in that activate the enzyme, phospholipase C (PLC), that cleaves they form the lipid bilayer that maintains cell integrity. -

Anti-Dicer (SAB4200087)
Anti-Dicer produced in rabbit, affinity isolated antibody Product Number SAB4200087 Product Description Precautions and Disclaimer Anti-Dicer is produced in rabbit using as the For R&D use only. Not for drug, household, or other immunogen a synthetic peptide corresponding to a uses. Please consult the Safety Data Sheet for fragment of human Dicer (Gene ID: 23405) conjugated information regarding hazards and safe handling to KLH. The corresponding sequence is identical in practices. mouse. The antibody is affinity-purified using the immunizing peptide immobilized on agarose. Storage/Stability Store at –20 C. For continuous use, store at 2–8 C for Anti-Dicer recognizes human Dicer. The antibody may up to one month. For extended storage, freeze in be used in several immunochemical techniques working aliquots at –20 C. Repeated freezing and including immunoblotting (218 kDa), immuno- thawing, or storage in “frost-free” freezers, is not precipitation, and immunofluorescence. Detection of recommended. If slight turbidity occurs upon prolonged the Dicer band by immunoblotting is specifically storage, clarify the solution by centrifugation before inhibited with the immunizing peptide. use. Working dilutions should be discarded if not used within 12 hours. Dicer, also known as Dicer1, Endoribonuclease Dicer, Helicase with RNase motif, and HERNA, is a member Product Profile of the RNase III family that catalyzes the first step in the Immunoblotting: a working antibody concentration of RNA interference (RNAi) pathway and initiates 3-6 g/mL is recommended using HeLa cell lysates. formation of the RNA-induced silencing complex (RISC). Dicer processes the dsRNA into small Immunoprecipitation: a working antibody amount of fragments called short interfering RNA (siRNA) or 2.5-5 g is recommended using HeLa cell lysates. -
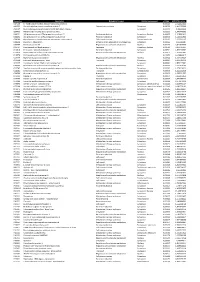
Accession Description Biological Process Cellular Component P-Value SCZ/CTRL Ration A4FU69 EF-Hand Calcium-Binding Domain-Contai
Accession Description Biological Process Cellular component p-Value SCZ/CTRL ration A4FU69 EF-hand calcium-binding domain-containing protein 5 0,001101 2,724427411 A4UGR9 Xin actin-binding repeat-containing protein 2 Cytoskeletal anchoring Cytoplasm 0,006756 1,413953388 A6NCE7 Microtubule-associated proteins 1A/1B light chain 3 beta 2 Cytoplasm 0,001417 1,99612969 B2RPK0 Putative high mobility group protein B1-like 1 0,032314 1,590743352 O00231 26S proteasome non-ATPase regulatory subunit 11 Protein metabolism Cytoplasm; Nucleus 0,000029 1,428664042 O00232 26S proteasome non-ATPase regulatory subunit 12 Protein metabolism Cytoplasm 0,008566 1,544545922 O00264 Membrane-associated progesterone receptor component 1 Cell communication Plasma membrane 0,001459 2,322924147 O00429 Dynamin-1-like protein Mitochondrion organization and biogenesis Cytoplasm 0,006560 0,06391487 O00567 Nucleolar protein 56 Regulation of nucleotide metabolism Nucleus 0,007330 3,842896338 O14737 Programmed cell death protein 5 Apoptosis Cytoplasm; Nucleus 0,006358 3,836727237 O14818 Proteasome subunit alpha type-7 Protein metabolism Cytoplasm 0,030521 1,893928387 O14979 Heterogeneous nuclear ribonucleoprotein D-like Regulation of nucleotide metabolism Nucleus 0,000637 2,150005885 O15078 Centrosomal protein of 290 kDa 0,015359 14,26648619 O15347 High mobility group protein B3 Regulation of nucleotide metabolism Nucleus 0,005500 1,364309014 O15540 Fatty acid-binding protein_ brain Transport Cytoplasm 0,000087 3,125786118 O43237 Cytoplasmic dynein 1 light intermediate -

The Structure-Function Relationship of Angular Estrogens and Estrogen Receptor Alpha to Initiate Estrogen-Induced Apoptosis in Breast Cancer Cells S
Supplemental material to this article can be found at: http://molpharm.aspetjournals.org/content/suppl/2020/05/03/mol.120.119776.DC1 1521-0111/98/1/24–37$35.00 https://doi.org/10.1124/mol.120.119776 MOLECULAR PHARMACOLOGY Mol Pharmacol 98:24–37, July 2020 Copyright ª 2020 The Author(s) This is an open access article distributed under the CC BY Attribution 4.0 International license. The Structure-Function Relationship of Angular Estrogens and Estrogen Receptor Alpha to Initiate Estrogen-Induced Apoptosis in Breast Cancer Cells s Philipp Y. Maximov, Balkees Abderrahman, Yousef M. Hawsawi, Yue Chen, Charles E. Foulds, Antrix Jain, Anna Malovannaya, Ping Fan, Ramona F. Curpan, Ross Han, Sean W. Fanning, Bradley M. Broom, Daniela M. Quintana Rincon, Jeffery A. Greenland, Geoffrey L. Greene, and V. Craig Jordan Downloaded from Departments of Breast Medical Oncology (P.Y.M., B.A., P.F., D.M.Q.R., J.A.G., V.C.J.) and Computational Biology and Bioinformatics (B.M.B.), University of Texas, MD Anderson Cancer Center, Houston, Texas; King Faisal Specialist Hospital and Research (Gen.Org.), Research Center, Jeddah, Kingdom of Saudi Arabia (Y.M.H.); The Ben May Department for Cancer Research, University of Chicago, Chicago, Illinois (R.H., S.W.F., G.L.G.); Center for Precision Environmental Health and Department of Molecular and Cellular Biology (C.E.F.), Mass Spectrometry Proteomics Core (A.J., A.M.), Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Mass Spectrometry Proteomics Core (A.M.), and Dan L. Duncan molpharm.aspetjournals.org -

The Regulatory Roles of Phosphatases in Cancer
Oncogene (2014) 33, 939–953 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW The regulatory roles of phosphatases in cancer J Stebbing1, LC Lit1, H Zhang, RS Darrington, O Melaiu, B Rudraraju and G Giamas The relevance of potentially reversible post-translational modifications required for controlling cellular processes in cancer is one of the most thriving arenas of cellular and molecular biology. Any alteration in the balanced equilibrium between kinases and phosphatases may result in development and progression of various diseases, including different types of cancer, though phosphatases are relatively under-studied. Loss of phosphatases such as PTEN (phosphatase and tensin homologue deleted on chromosome 10), a known tumour suppressor, across tumour types lends credence to the development of phosphatidylinositol 3--kinase inhibitors alongside the use of phosphatase expression as a biomarker, though phase 3 trial data are lacking. In this review, we give an updated report on phosphatase dysregulation linked to organ-specific malignancies. Oncogene (2014) 33, 939–953; doi:10.1038/onc.2013.80; published online 18 March 2013 Keywords: cancer; phosphatases; solid tumours GASTROINTESTINAL MALIGNANCIES abs in sera were significantly associated with poor survival in Oesophageal cancer advanced ESCC, suggesting that they may have a clinical utility in Loss of PTEN (phosphatase and tensin homologue deleted on ESCC screening and diagnosis.5 chromosome 10) expression in oesophageal cancer is frequent, Cao et al.6 investigated the role of protein tyrosine phosphatase, among other gene alterations characterizing this disease. Zhou non-receptor type 12 (PTPN12) in ESCC and showed that PTPN12 et al.1 found that overexpression of PTEN suppresses growth and protein expression is higher in normal para-cancerous tissues than induces apoptosis in oesophageal cancer cell lines, through in 20 ESCC tissues. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds.