Prioritization and Comprehensive Analysis of Genes Associated with Melanoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MAPK1/MAPK3 Monoclonal Antibody, Clone Sb46b
MAPK1/MAPK3 monoclonal antibody, clone SB46b Catalog # : MAB3337 規格 : [ 500 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against recombinant Western Blot Description: MAPK1/MAPK3. Immunogen: Recombinant protein corresponding to human MAPK1/MAPK3. Host: Mouse enlarge Reactivity: Human Immunoprecipitation Specificity: human Erk 1 and human Erk2. Form: Liquid Isotype: IgG2a Recommend Western Blot (1-2 ug/mL) Usage: Immunoprecipitation (10-15ug) The optimal working dilution should be determined by the end user. Storage Buffer: In 100 mM BBS, pH 8.0 Storage Store at 4°C. Instruction: Datasheet: Download Publication Reference 1. Hydrophobic as well as charged residues in both MEK1 and ERK2 are important for their proper docking. Xu Be, Stippec S, Robinson FL, Cobb MH.J Biol Chem. 2001 Jul 13;276(28):26509-15. Epub 2001 May 14. 2. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH.Endocr Rev. 2001 Apr;22(2):153-83. 3. Molecular cloning, expression, and characterization of the human mitogen-activated protein kinase p44erk1. Charest DL, Mordret G, Harder KW, Jirik F, Pelech SL.Mol Cell Biol. 1993 Aug;13(8):4679-90. Applications Western Blot Page 1 of 3 2021/6/20 Human PBMC (Lane 1) and spleen lysate (Lane 2) was stained with MAPK1/3 monoclonal antibody, clone SB46b (Cat # MAB3337). Goat anti-Mouse IgG2a was used to develop. Immunoprecipitation MAPK1 MAPK3 Gene Information Entrez GeneID: 5595 Gene Name: MAPK3 Gene Alias: ERK1,HS44KDAP,HUMKER1A,MGC20180,P44ERK1,P44MAPK,PRKM3 Gene mitogen-activated protein kinase 3 Description: Omim ID: 601795 Gene Ontology: Hyperlink Gene Summary: The protein encoded by this gene is a member of the MAP kinase family. -

N-Glycan Trimming in the ER and Calnexin/Calreticulin Cycle
Neurotransmitter receptorsGABA and A postsynapticreceptor activation signal transmission Ligand-gated ion channel transport GABAGABA Areceptor receptor alpha-5 alpha-1/beta-1/gamma-2 subunit GABA A receptor alpha-2/beta-2/gamma-2GABA receptor alpha-4 subunit GABAGABA receptor A receptor beta-3 subunitalpha-6/beta-2/gamma-2 GABA-AGABA receptor; A receptor alpha-1/beta-2/gamma-2GABA receptoralpha-3/beta-2/gamma-2 alpha-3 subunit GABA-A GABAreceptor; receptor benzodiazepine alpha-6 subunit site GABA-AGABA-A receptor; receptor; GABA-A anion site channel (alpha1/beta2 interface) GABA-A receptor;GABA alpha-6/beta-3/gamma-2 receptor beta-2 subunit GABAGABA receptorGABA-A receptor alpha-2receptor; alpha-1 subunit agonist subunit GABA site Serotonin 3a (5-HT3a) receptor GABA receptorGABA-C rho-1 subunitreceptor GlycineSerotonin receptor subunit3 (5-HT3) alpha-1 receptor GABA receptor rho-2 subunit GlycineGlycine receptor receptor subunit subunit alpha-2 alpha-3 Ca2+ activated K+ channels Metabolism of ingested SeMet, Sec, MeSec into H2Se SmallIntermediateSmall conductance conductance conductance calcium-activated calcium-activated calcium-activated potassium potassium potassiumchannel channel protein channel protein 2 protein 1 4 Small conductance calcium-activatedCalcium-activated potassium potassium channel alpha/beta channel 1 protein 3 Calcium-activated potassiumHistamine channel subunit alpha-1 N-methyltransferase Neuraminidase Pyrimidine biosynthesis Nicotinamide N-methyltransferase Adenosylhomocysteinase PolymerasePolymeraseHistidine basic -

Anti-EGFR Monoclonal Antibodies and EGFR Tyrosine Kinase Inhibitors As Combination Therapy for Triple-Negative Breast Cancer
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 45 Research Paper Anti-EGFR monoclonal antibodies and EGFR tyrosine kinase inhibitors as combination therapy for triple-negative breast cancer Abderrahim El Guerrab1,2, Mahchid Bamdad2,3, Fabrice Kwiatkowski1, Yves-Jean Bignon1,2,*, Frédérique Penault-Llorca1,2,*, Corinne Aubel1,2 1Centre Jean Perrin - ERTICa-EA4677, BP392, 63011 Clermont-Ferrand Cedex, France 2Clermont Université - Université d’Auvergne - ERTICa-EA4677, Faculté de Médecine, BP38, 63001 Clermont-Ferrand Cedex, France 3Clermont Université - Université d’Auvergne - ERTICa-EA4677, Institut Universitaire de Technologie, Département Génie Biologique, Ensemble Universitaire des Cézeaux, BP86, 63172 Aubière Cedex, France *These authors have contributed equally to this work Correspondence to: Yves-Jean Bignon, email: [email protected] Keywords: triple-negative breast cancer, epidermal growth factor receptor, anti-EGFR targeted therapy, cytotoxicity, cell cycle Received: November 09, 2015 Accepted: August 22, 2016 Published: September 15, 2016 ABSTRACT Triple-negative breast cancer (TNBC) is characterized by overexpression of epidermal growth factor receptor (EGFR) and activation of its downstream signaling pathways. Dual targeting of EGFR using one monoclonal antibody (mAb; cetuximab or panitumumab) and one tyrosine kinase inhibitor (EGFR-TKI; gefitinib or erlotinib) is a potential therapeutic approach. We investigated the effect of these therapies in EGFR-expressing TNBC cell lines that do or do not harbor the main activating mutations of EGFR pathways. Cell lines were sensitive to EGFR-TKIs, whereas mAbs were active only in MDA-MB-468 (EGFR amplification) and SUM-1315 (KRAS and PTEN wild-type) cells. MDA-MB-231 (KRAS mutated) and HCC-1937 (PTEN deletion) cells were resistant to mAbs. -

Modulation of NF-Κb Signalling by Microbial Pathogens
REVIEWS Modulation of NF‑κB signalling by microbial pathogens Masmudur M. Rahman and Grant McFadden Abstract | The nuclear factor-κB (NF‑κB) family of transcription factors plays a central part in the host response to infection by microbial pathogens, by orchestrating the innate and acquired host immune responses. The NF‑κB proteins are activated by diverse signalling pathways that originate from many different cellular receptors and sensors. Many successful pathogens have acquired sophisticated mechanisms to regulate the NF‑κB signalling pathways by deploying subversive proteins or hijacking the host signalling molecules. Here, we describe the mechanisms by which viruses and bacteria micromanage the host NF‑κB signalling circuitry to favour the continued survival of the pathogen. The nuclear factor-κB (NF-κB) family of transcription Signalling targets upstream of NF‑κB factors regulates the expression of hundreds of genes that NF-κB proteins are tightly regulated in both the cyto- are associated with diverse cellular processes, such as pro- plasm and the nucleus6. Under normal physiological liferation, differentiation and death, as well as innate and conditions, NF‑κB complexes remain inactive in the adaptive immune responses. The mammalian NF‑κB cytoplasm through a direct interaction with proteins proteins are members of the Rel domain-containing pro- of the inhibitor of NF-κB (IκB) family, including IκBα, tein family: RELA (also known as p65), RELB, c‑REL, IκBβ and IκBε (also known as NF-κBIα, NF-κBIβ and the NF-κB p105 subunit (also known as NF‑κB1; which NF-κBIε, respectively); IκB proteins mask the nuclear is cleaved into the p50 subunit) and the NF-κB p100 localization domains in the NF‑κB complex, thus subunit (also known as NF‑κB2; which is cleaved into retaining the transcription complex in the cytoplasm. -

Supplementary Table 1. in Vitro Side Effect Profiling Study for LDN/OSU-0212320. Neurotransmitter Related Steroids
Supplementary Table 1. In vitro side effect profiling study for LDN/OSU-0212320. Percent Inhibition Receptor 10 µM Neurotransmitter Related Adenosine, Non-selective 7.29% Adrenergic, Alpha 1, Non-selective 24.98% Adrenergic, Alpha 2, Non-selective 27.18% Adrenergic, Beta, Non-selective -20.94% Dopamine Transporter 8.69% Dopamine, D1 (h) 8.48% Dopamine, D2s (h) 4.06% GABA A, Agonist Site -16.15% GABA A, BDZ, alpha 1 site 12.73% GABA-B 13.60% Glutamate, AMPA Site (Ionotropic) 12.06% Glutamate, Kainate Site (Ionotropic) -1.03% Glutamate, NMDA Agonist Site (Ionotropic) 0.12% Glutamate, NMDA, Glycine (Stry-insens Site) 9.84% (Ionotropic) Glycine, Strychnine-sensitive 0.99% Histamine, H1 -5.54% Histamine, H2 16.54% Histamine, H3 4.80% Melatonin, Non-selective -5.54% Muscarinic, M1 (hr) -1.88% Muscarinic, M2 (h) 0.82% Muscarinic, Non-selective, Central 29.04% Muscarinic, Non-selective, Peripheral 0.29% Nicotinic, Neuronal (-BnTx insensitive) 7.85% Norepinephrine Transporter 2.87% Opioid, Non-selective -0.09% Opioid, Orphanin, ORL1 (h) 11.55% Serotonin Transporter -3.02% Serotonin, Non-selective 26.33% Sigma, Non-Selective 10.19% Steroids Estrogen 11.16% 1 Percent Inhibition Receptor 10 µM Testosterone (cytosolic) (h) 12.50% Ion Channels Calcium Channel, Type L (Dihydropyridine Site) 43.18% Calcium Channel, Type N 4.15% Potassium Channel, ATP-Sensitive -4.05% Potassium Channel, Ca2+ Act., VI 17.80% Potassium Channel, I(Kr) (hERG) (h) -6.44% Sodium, Site 2 -0.39% Second Messengers Nitric Oxide, NOS (Neuronal-Binding) -17.09% Prostaglandins Leukotriene, -

Combined Inhibition of MEK and Plk1 Has Synergistic Anti-Tumor Activity in NRAS Mutant Melanoma
Combined inhibition of MEK and Plk1 has synergistic anti-tumor activity in NRAS mutant melanoma The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Posch, C., B. Cholewa, I. Vujic, M. Sanlorenzo, J. Ma, S. Kim, S. Kleffel, et al. 2015. “Combined inhibition of MEK and Plk1 has synergistic anti-tumor activity in NRAS mutant melanoma.” The Journal of investigative dermatology 135 (10): 2475-2483. doi:10.1038/jid.2015.198. http://dx.doi.org/10.1038/jid.2015.198. Published Version doi:10.1038/jid.2015.198 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26860102 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA HHS Public Access Author manuscript Author Manuscript Author ManuscriptJ Invest Author ManuscriptDermatol. Author Author Manuscript manuscript; available in PMC 2016 April 01. Published in final edited form as: J Invest Dermatol. 2015 October ; 135(10): 2475–2483. doi:10.1038/jid.2015.198. Combined inhibition of MEK and Plk1 has synergistic anti-tumor activity in NRAS mutant melanoma C Posch#1,2,3,*, BD Cholewa#4, I Vujic1,3, M Sanlorenzo1,5, J Ma1, ST Kim1, S Kleffel2, T Schatton2, K Rappersberger3, R Gutteridge4, N Ahmad4, and S Ortiz/Urda1 1 University of California San Francisco, Department of Dermatology, Mt. Zion Cancer Research -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach
Cancers 2019, 11, x S1 of S9 Supplementary Materials: Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach Young Rae Kim, Yong Wan Kim, Suh Eun Lee, Hye Won Yang and Sung Young Kim Table S1. Characteristics of individual studies. Sample Size Origin of Cancer Drug Dataset Platform S AR (Cell Lines) Lung cancer Agilent-014850 Whole Human GSE34228 26 26 (PC9) Genome Microarray 4x44K Gefitinib Epidermoid carcinoma Affymetrix Human Genome U133 GSE10696 3 3 (A431) Plus 2.0 Head and neck cancer Illumina HumanHT-12 V4.0 GSE62061 12 12 (Cal-27, SSC-25, FaDu, expression beadchip SQ20B) Erlotinib Head and neck cancer Illumina HumanHT-12 V4.0 GSE49135 3 3 (HN5) expression beadchip Lung cancer (HCC827, Illumina HumanHT-12 V3.0 GSE38310 3 6 ER3, T15-2) expression beadchip Lung cancer Illumina HumanHT-12 V3.0 GSE62504 1 2 (HCC827) expression beadchip Afatinib Lung cancer * Illumina HumanHT-12 V4.0 GSE75468 1 3 (HCC827) expression beadchip Head and neck cancer Affymetrix Human Genome U133 Cetuximab GSE21483 3 3 (SCC1) Plus 2.0 Array GEO, gene expression omnibus; GSE, gene expression series; S, sensitive; AR, acquired EGFR-TKI resistant; * Lung Cancer Cells Derived from Tumor Xenograft Model. Table S2. The performances of four penalized regression models. Model ACC precision recall F1 MCC AUROC BRIER Ridge 0.889 0.852 0.958 0.902 0.782 0.964 0.129 Lasso 0.944 0.957 0.938 0.947 0.889 0.991 0.042 Elastic Net 0.978 0.979 0.979 0.979 0.955 0.999 0.023 EPSGO Elastic Net 0.989 1.000 0.979 0.989 0.978 1.000 0.018 AUROC, area under curve of receiver operating characteristic; ACC, accuracy; MCC, Matthews correlation coefficient; EPSGO, Efficient Parameter Selection via Global Optimization algorithm. -

Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in Vitro and in Vivo
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Genetik Characterization of the Small Molecule Kinase Inhibitor SU11248 (Sunitinib/ SUTENT in vitro and in vivo - Towards Response Prediction in Cancer Therapy with Kinase Inhibitors Michaela Bairlein Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ. -Prof. Dr. K. Schneitz Prüfer der Dissertation: 1. Univ.-Prof. Dr. A. Gierl 2. Hon.-Prof. Dr. h.c. A. Ullrich (Eberhard-Karls-Universität Tübingen) 3. Univ.-Prof. A. Schnieke, Ph.D. Die Dissertation wurde am 07.01.2010 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 19.04.2010 angenommen. FOR MY PARENTS 1 Contents 2 Summary ................................................................................................................................................................... 5 3 Zusammenfassung .................................................................................................................................................... 6 4 Introduction .............................................................................................................................................................. 8 4.1 Cancer .............................................................................................................................................................. -
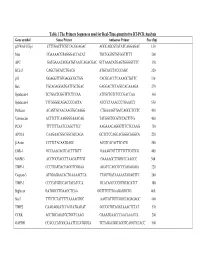
Table 1 the Primers Sequences Used for Real-Time Quantitative RT-PCR
Table 1 The Primers Sequences used for Real-Time quantitative RT-PCR Analysis Gene symbol Sense Primer Antisense Primer Size (bp) p21WAF1/Cip1 CTTTGACTTCGTCACGGAGAC AGGCAGCGTATATCAGGAGAC 150 Max CGAAAACGTAGGGACCACAT TGCTGGTGTGTGGTTTTT 200 APC GATGAAATAGGATGTAATCAGACGAC GCTAAACATGAGTGGGGTCTC 350 BCL-2 CAGCTGCACCTGACG ATGCACCTACCCAGC 320 p53 GGAGGTTGTGAGGCGCTGG CACGCACCTCAAAGCTGTTC 330 Bax TGCAGAGGATGATTGCTGAC GAGGACTCCAGCCACAAAGA 270 Syndecan-1 TCTGACTCGGTTTCTCCAA ATTGCTGTCTCCCGACCAA 360 Syndecan-3 TTTGGGGCAGACCCCACTA ATCCCTAAACCCTGAACCT 550 Perlecan ACAGTGCAACAAGTGCAAGG CTGAAAGTGACCAGGCTCCTC 450 Vitronectin ACTTCTTCAAGGGGAAACAG TATGGCTGCGTCCACTTTG 400 PCAF TTTTTTTAATCCAGCTTCC AAGAAACAGGGTTTCTCCAAG 750 APO1A CAAGAACGGCGGCGCCAGA GCTCTCCAGCACGGGCAGGCA 220 β-Actin CCTTCTACAATGAGC ACGTCACACTTCATG 260 EGR-1 GCCAAACAGTCACTTTGTT GAAAGTGTTTTTTCTTCGTCG 400 MAPK3 ACCTGCGACCTTAAGATTTGT GAAAAGCTTGGCCCAAGCC 300 TIMP-1 CCCTGATGACGAGGTCGGAA AGATCCAGCGCCCAGAGAGA 220 Caspase-3 ATGGAGAACACTGAAAACTCA TTAGTGATAAAAATAGAGTTC 200 TIMP-3 CCCCATGTGCAGTACATCCA GCACAGCCCCGTGTACATCT 180 Biglycan GATGGCCTGAAGCTCAA GGTTTGTTGAAGAGGCTG 460 Stat3 TTTCTCTATTTTTAAAAGTGC AAGTATTGTCGGCCAGAGAGC 460 TIMP2 CAAGAGGATCCAGTATGAGAT GCCCGTGTAGATAAACTCTAT 570 CCRK ACCTGCAGATGCTGCTCAAG GAAATGAACCCAACAAACCA 200 GAPDH CCACCCATGGCAAATTCCATGGGA TCTAGACGGCAGGTCAGGTCCACC 500 Table 2. Genes with Altered Expression in Cleaved vs. Native Antithrombin-Treated HUVECs Gene Fold Changes Gene Symbol Gene Description Cluster ID (ATC/ATN) Adhesion/ECM ARGN Agrin Hs.273330 2.18 CDH24 Caderhrin-like -

Activation of Diverse Signalling Pathways by Oncogenic PIK3CA Mutations
ARTICLE Received 14 Feb 2014 | Accepted 12 Aug 2014 | Published 23 Sep 2014 DOI: 10.1038/ncomms5961 Activation of diverse signalling pathways by oncogenic PIK3CA mutations Xinyan Wu1, Santosh Renuse2,3, Nandini A. Sahasrabuddhe2,4, Muhammad Saddiq Zahari1, Raghothama Chaerkady1, Min-Sik Kim1, Raja S. Nirujogi2, Morassa Mohseni1, Praveen Kumar2,4, Rajesh Raju2, Jun Zhong1, Jian Yang5, Johnathan Neiswinger6, Jun-Seop Jeong6, Robert Newman6, Maureen A. Powers7, Babu Lal Somani2, Edward Gabrielson8, Saraswati Sukumar9, Vered Stearns9, Jiang Qian10, Heng Zhu6, Bert Vogelstein5, Ben Ho Park9 & Akhilesh Pandey1,8,9 The PIK3CA gene is frequently mutated in human cancers. Here we carry out a SILAC-based quantitative phosphoproteomic analysis using isogenic knockin cell lines containing ‘driver’ oncogenic mutations of PIK3CA to dissect the signalling mechanisms responsible for oncogenic phenotypes induced by mutant PIK3CA. From 8,075 unique phosphopeptides identified, we observe that aberrant activation of PI3K pathway leads to increased phosphorylation of a surprisingly wide variety of kinases and downstream signalling networks. Here, by integrating phosphoproteomic data with human protein microarray-based AKT1 kinase assays, we discover and validate six novel AKT1 substrates, including cortactin. Through mutagenesis studies, we demonstrate that phosphorylation of cortactin by AKT1 is important for mutant PI3K-enhanced cell migration and invasion. Our study describes a quantitative and global approach for identifying mutation-specific signalling events and for discovering novel signalling molecules as readouts of pathway activation or potential therapeutic targets. 1 McKusick-Nathans Institute of Genetic Medicine and Department of Biological Chemistry, Johns Hopkins University School of Medicine, 733 North Broadway, BRB 527, Baltimore, Maryland 21205, USA. -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain