The Case of Bitter Taste Receptors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

G Protein-Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. British Journal of Pharmacology (2015) 172, 5744–5869 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: G protein-coupled receptors Stephen PH Alexander1, Anthony P Davenport2, Eamonn Kelly3, Neil Marrion3, John A Peters4, Helen E Benson5, Elena Faccenda5, Adam J Pawson5, Joanna L Sharman5, Christopher Southan5, Jamie A Davies5 and CGTP Collaborators 1School of Biomedical Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK, 2Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK, 3School of Physiology and Pharmacology, University of Bristol, Bristol, BS8 1TD, UK, 4Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK, 5Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2015/16 provides concise overviews of the key properties of over 1750 human drug targets with their pharmacology, plus links to an open access knowledgebase of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. The full contents can be found at http://onlinelibrary.wiley.com/doi/ 10.1111/bph.13348/full. G protein-coupled receptors are one of the eight major pharmacological targets into which the Guide is divided, with the others being: ligand-gated ion channels, voltage-gated ion channels, other ion channels, nuclear hormone receptors, catalytic receptors, enzymes and transporters. These are presented with nomenclature guidance and summary information on the best available pharmacological tools, alongside key references and suggestions for further reading. -

Intracellular Zinc-Dependent TAS2R8 Gene Expression Through CTCF Activa- Tion
Biomedical Research (Tokyo) 41 (5) 217–225, 2020 Intracellular zinc-dependent TAS2R8 gene expression through CTCF activa- tion 1 1, 2 2 1, 3 2, 4 Tsuyoshi KOJIMA , Toyonobu MAEDA , Atsuko SUZUKI , Tetsuo YAMAMORI , and Yasumasa KATO Departments of 1 Oral Rehabilitation and 4 Oral Physiology and Biochemistry, Ohu University Graduate School of Dentistry, Koriyama 963-8611, Japan; Departments of 2 Oral Function and Molecular Biology and 3 Prosthetic Dentistry, Ohu University School of Dentistry, Koriyama 963-8611, Japan (Received 21 April 2020; and accepted 7 June 2020) ABSTRACT Taste-2 receptors (TAS2Rs), which belong to the G-protein coupled receptor (GPCR) family, are receptors for bitter taste perception. The aim of this study was to investigate whether zinc defi- ciency affects the expression of TAS2R genes. The promoter activity of the TAS2R7, TAS2R8, and TAS2R42 genes were determined in Ca9-22 oral squamous cell carcinoma cells cultured in the presence or absence of zinc. Luciferase reporter assays showed that zinc deprivation inhibited TAS2R8 promoter activity, but not the promoter activity of the other two genes. Treatment of the cells with N,N,N’,N’-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN), an intracellular chela- tor of Zn2+, in the presence of 10% fetal bovine serum reduced TAS2R8 promoter activity. Trunca- tion/deletion mutants of TAS2R8 promoter-luciferase constructs showed that the region from nucleotide −1152 to nucleotide −925 was critical for intracellular zinc dependency and contained a CCCTC-binding factor (CTCF) binding motif. A chromatin immunoprecipitation (ChiP) assay showed that CTCF bound specifically to this region, a binding abrogated by zinc deficiency, sug- gesting that CTCF plays a critical role in zinc-dependent bitter taste perception through TAS2R8. -

G Protein‐Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: G protein-coupled receptors. British Journal of Pharmacology (2019) 176, S21–S141 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors Stephen PH Alexander1 , Arthur Christopoulos2 , Anthony P Davenport3 , Eamonn Kelly4, Alistair Mathie5 , John A Peters6 , Emma L Veale5 ,JaneFArmstrong7 , Elena Faccenda7 ,SimonDHarding7 ,AdamJPawson7 , Joanna L Sharman7 , Christopher Southan7 , Jamie A Davies7 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Monash Institute of Pharmaceutical Sciences and Department of Pharmacology, Monash University, Parkville, Victoria 3052, Australia 3Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK 4School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 5Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 6Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 7Centre for Discovery Brain Sciences, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. Although the Concise Guide represents approximately 400 pages, the material presented is substantially reduced compared to information and links presented on the website. -

Origins and Adaptation in Humans: a Case Study of Taste and Lifestyle
Origins and adaptation in humans : a case study of taste and lifestyle Agnès Sjöstrand To cite this version: Agnès Sjöstrand. Origins and adaptation in humans : a case study of taste and lifestyle. Human genetics. Université Pierre et Marie Curie - Paris VI; Uppsala universitet, 2015. English. NNT : 2015PA066724. tel-01609900 HAL Id: tel-01609900 https://tel.archives-ouvertes.fr/tel-01609900 Submitted on 4 Oct 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Université Pierre et Marie Curie Université d’Uppsala ED227 : Sciences de la Nature et de l’Homme : Écologie et Évolution UMR7206 Eco-Anthropologie et Ethnobiologie, Evolutionary Biology Center (Uppsala University) UMR5525 Laboratoire TIMC-IMAG. Origins and Adaptation in Humans a Case Study of Taste and Lifestyle Par Agnès Sjöstrand Thèse de doctorat de Génétique des populations Dirigée par Pr. Evelyne Heyer, Pr. Mattias Jakobsson & Dr. Michael Blum Présentée et soutenue publiquement le 20 novembre 2015 Devant un jury composé de : David Comas, professeur (rapporteur) Luis Quintana-Murci, directeur de recherche (rapporteur) Thierry Wirth, professeur (examinateur) Denis Pierron, chargé de recherche (examinateur) Åsa Johansson, chercheuse (examinatrice) Evelyne Heyer, professeure (directrice de thèse) Mattias Jakobsson, professeur (directeur de thèse) Michaël Blum, directeur de recherche (co-directeur de thèse) Ubuntu (Nguni word) Shosholoza (Ndebele word) List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. -

Genetic Variations in TAS2R3 and TAS2R4 Bitterness Receptors Modify
www.nature.com/scientificreports OPEN Genetic variations in TAS2R3 and TAS2R4 bitterness receptors modify papillary carcinoma risk and Received: 10 January 2018 Accepted: 25 September 2018 thyroid function in Korean females Published: xx xx xxxx Jeong-Hwa Choi 1,2, Jeonghee Lee 1, Sarah Yang1,3, Eun Kyung Lee4, Yul Hwangbo4 & Jeongseon Kim 1 Type 2 taste receptors (T2Rs, TAS2Rs) mediate bitterness perception and are involved in diverse defence mechanisms in extraoral tissues. The thyrocyte-expressed T2Rs control thyroid hormone production, and this regulatory role may be associated with susceptibility to thyroid diseases. This study examined whether the variations in TAS2Rs modify the risk of papillary thyroid carcinoma (PTC) and whether such T2R-related PTC risk is associated with genetically modifed thyroid function. We conducted a case-control study with 763 Korean females, including 250 PTC cases. Seventy-three single-nucleotide polymorphisms in 13 TAS2R genes and the pre-diagnosis levels of 4 thyroid-related functional markers [total triiodothyronine (TT3), free thyroxine, thyroid-stimulating hormone and thyroglobulin] were analysed. Individuals with TAS2R3/4 CC haplotype (rs2270009 and rs2234001) were at a lower risk for PTC than those with the remaining haplotypes (odds ratio = 0.59, 95% confdence interval: 0.36–0.97). Furthermore, TT3 levels were signifcantly reduced for TAS2R3/4 CC haplotype carriers compared with other haplotype carriers (p = 0.005). No other genetic variants exhibited critical associations with the PTC phenotype and biomarkers. In summary, genetic variations in T2R3/4 bitterness receptors may modify the PTC risk, and the genetically modifed thyroid hormone level by those variations may be linked with the PTC-T2Rs association. -

Oxygenated Fatty Acids Enhance Hematopoiesis Via the Receptor GPR132
Oxygenated Fatty Acids Enhance Hematopoiesis via the Receptor GPR132 The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Lahvic, Jamie L. 2017. Oxygenated Fatty Acids Enhance Hematopoiesis via the Receptor GPR132. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:42061504 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Oxygenated Fatty Acids Enhance Hematopoiesis via the Receptor GPR132 A dissertation presented by Jamie L. Lahvic to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Developmental and Regenerative Biology Harvard University Cambridge, Massachusetts May 2017 © 2017 Jamie L. Lahvic All rights reserved. Dissertation Advisor: Leonard I. Zon Jamie L. Lahvic Oxygenated Fatty Acids Enhance Hematopoiesis via the Receptor GPR132 Abstract After their specification in early development, hematopoietic stem cells (HSCs) maintain the entire blood system throughout adulthood as well as upon transplantation. The processes of HSC specification, renewal, and homing to the niche are regulated by protein, as well as lipid signaling molecules. A screen for chemical enhancers of marrow transplant in the zebrafish identified the endogenous lipid signaling molecule 11,12-epoxyeicosatrienoic acid (11,12-EET). EET has vasodilatory properties, but had no previously described function on HSCs. -

Article T2R Bitter Taste Receptors Regulate Apoptosis and May Be
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.17.444527; this version posted May 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. T2R-mediated apoptosis in HNSCC Article T2R bitter taste receptors regulate apoptosis and may be associated with survival in head and neck squamous cell carcinoma RUNNING TITLE: T2R-mediated apoptosis in HNSCC Ryan M. Carey,1,* Derek B. McMahon,1 Karthik Rajasekaran,1 Indiwari Gopallawa,1 Jason G. Newman,1 Devraj Basu,1 Elizabeth A. White,1 and Robert J. Lee1,2,* 1Department of Otorhinolaryngology, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania, USA 2Department of Physiology, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania, USA *To whom correspondence should be addressed: Ryan M. Carey or Robert J. Lee, Department of Otorhinolaryngology—Head and Neck Surgery, Hospital of the University of Pennsylvania, 3400 Spruce Street, 5th floor Ravdin Suite A, Philadelphia, PA 19104; [email protected] (R.M.C.) or [email protected] (R.J.L.). tel: 215-573-9766 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.05.17.444527; this version posted May 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. T2R-mediated apoptosis in HNSCC Abstract Better management of head and neck squamous cell carcinomas (HNSCCs) requires a clearer understanding of tumor biology and disease risk. Bitter taste receptors (T2Rs) have been studied in several cancers, including thyroid, salivary, and GI, but their role in HNSCC has not been explored. -

An Integrated Analysis Reveals Geniposide Extracted from Gardenia Jasminoides Ellis Regulates Calcium Signaling Pathway Essential for Infuenza a Virus Replication
An Integrated Analysis Reveals Geniposide Extracted From Gardenia Jasminoides Ellis Regulates Calcium Signaling Pathway Essential for Inuenza a Virus Replication LiRun Zhou China Academy of Chinese Medical Sciences https://orcid.org/0000-0002-1350-940X Lei Bao China Academy of Chinese Medical Sciences Yaxin Wang China Academy of Chinese Medical Sciences Mengping Chen China Academy of Chinese Medical Sciences Yingying Zhang Beijing University of Chinese Medicine Aliated Dongzhimen Hospital Zihan Geng China Academy of Chinese Medical Sciences Ronghua Zhao China Academy of Chinese Medical Sciences Jing Sun China Academy of Chinese Medical Sciences Yanyan Bao China Academy of Chinese Medical Sciences Yujing Shi China Academy of Chinese Medical Sciences Rongmei Yao China Academy of Chinese Medical Sciences Shanshan Guo ( [email protected] ) China Academy of Chinese Medical Sciences Xiaolan Cui China Academy of Chinese Medical Sciences Research Page 1/30 Keywords: Geniposide, Inuenza A virus, RNA polymerase, Calcium signaling pathway, CAMKII Posted Date: August 9th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-764303/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/30 Abstract Background Geniposide, an iridoid glycoside puried from the fruit of Gardenia jasminoides Ellis, has been reported to possess pleiotropic activity against different diseases. In particular, geniposide possesses a variety of biological activities and exerts good therapeutic effects in the treatment of several strains of the inuenza virus. However, the molecular mechanism for the therapeutic effect has not been well dened. Methods This study aimed to investigate the mechanism of geniposide on inuenza A virus (IAV). -
Selectivity Determinants of GPCR–G-Protein Binding Tilman Flock1,2, Alexander S
ARTICLE doi:10.1038/nature22070 Selectivity determinants of GPCR–G-protein binding Tilman Flock1,2, Alexander S. Hauser3, Nadia Lund3, David E. Gloriam3, Santhanam Balaji1 & M. Madan Babu1 The selective coupling of G-protein-coupled receptors (GPCRs) to specific G proteins is critical to trigger the appropriate physiological response. However, the determinants of selective binding have remained elusive. Here we reveal the existence of a selectivity barcode (that is, patterns of amino acids) on each of the 16 human G proteins that is recognized by distinct regions on the approximately 800 human receptors. Although universally conserved positions in the barcode allow the receptors to bind and activate G proteins in a similar manner, different receptors recognize the unique positions of the G-protein barcode through distinct residues, like multiple keys (receptors) opening the same lock (G protein) using non-identical cuts. Considering the evolutionary history of GPCRs allows the identification of these selectivity- determining residues. These findings lay the foundation for understanding the molecular basis of coupling selectivity within individual receptors and G proteins. Membrane protein receptors trigger the appropriate cellular response GPCR and Gα protein repertoires to extracellular stimuli by selective interaction with cytosolic adaptor Understanding how GPCRs and Gα proteins evolve could pro- proteins. In humans, GPCRs form the largest family of receptors, with vide insights into the constraints underlying selective coupling. over 800 members1–3. Although GPCRs bind a staggering number of The genomes of unicellular sister groups of metazoans (diverged natural ligands (~1,000), they primarily couple to only four major Gα ~900 million years ago) encode a small number of genes for the GPCR– families encoded by 16 human genes3,4. -

Supplementary Table S1 Demographic Characteristics Of
Supplementary Table S1 Demographic characteristics of HCC patients • Age (year; mean ± SD) 53.950 ± 11.802 • Gender (number; %) Male 79 (79%) Female 21 (21%) • AFP (ng/mL; mean ± SD) 23928.571 ± 79577.084 • Tumor encapsulation (number; %) Absent 61 (63.5%) Present 35 (36.5%) • Cellular differentiation by Edmondson grading I-II 46 (47.9%) III-IV 50 (52.1%) • Tumor microsatellite formation (number, %) Absent 45 (46.9%) Present 51 (53.1%) • Tumor size (cm; mean ± SD) Length 8.3 ± 5.0 Width 7.4 ± 4.3 • Venous invasion (number; %) Absent 42 (43.3%) Present 55 (56.7%) • Direct liver invasion (number; %) Absent 53 (60.2%) Present 35 (39.8%) • pTNM staging (number, %) I-II 37(38.1%) III-IV 60 (61.9%) • Hepatitis B surface antigen (number; %) Absent 22 (23.4%) Present 72 (76.6%) • Background liver disease (number; %) Normal 7 (7.2%) Chronic hepatitis 36 (37.1%) Cirrhosis 54 (55.7%) Supplementary Table S2 Primer sequences used in qRT-PCR study Genes Sequences Forward: cagatcatcggtcaggccaa Human NDUFA4 Reverse: tctgtcccaacaaacatctgga Forward: aaaagacatccggggatcat Human NDUFA4L2 Reverse: tccgggttgttctttctgtc Forward: ctcctccactgcccactcta Human NDUFA4L2 HRE-1 (ChIP) Reverse: gtcactctgtactctgcctt Forward: gagagcaggtccccagaga Human NDUFA4L2 HRE-2 (ChIP) Reverse: ctccttggcccagacttatg Forward: gaggatgaggtggaacgtgt Human 18S Reverse: agaagtgacgcagccctcta Forward: gcactcttccagccttcctt Human ACTB Reverse: aatgccagggtacatggtgg Forward: ctttgctgacctgctggatt Human HPRT Reverse: ctgcattgttttcggagtgt Supplementary Table S3 The target regions of shRNA and siRNA constructs shRNA clones Genebank Target nucleotides shNDUFA4-30 NM_002489 124-144 shNDUFA4-88 NM_002489 353-373 shNDUFA4L2-90 NM_020142 60-80 shHIF-1α NM_181054 2,123-2,141 shHIF-2α NM_001430 1,992-2,012 siRNA clones Genebank Target nucleotides siNDUFA4L2-1 NM_020142 246-264 siNDUFA4L2-3 NM_020142 103-121 siNDUFA4L2-4 NM_020142 146-164 Supplementary Table S4 Clinico-pathological correlation of NDUFA4L2 over-expression in human HCC Clinico-pathological features No. -

Nyctereutes Procyonoides)
Pakistan J. Zool., vol. 50(4), pp 1361-1366, 2018. DOI: http://dx.doi.org/10.17582/journal.pjz/2018.50.4.1361.1366 Supplementary Material The Bitter Taste Receptor Genes of the Raccoon Dog (Nyctereutes procyonoides) Shuai Shang1,2, Honghai Zhang2,*, Xiaoyang Wu2, Qinguo Wei2, Jun Chen1, Huanxin Zhang1, Huaming Zhong2 and Xuexi Tang1,* 1College of Marine Life, Ocean University of China, Qingdao, Shandong, China 2College of Life Science, Qufu Normal University, Qufu, Shandong, China * Corresponding authors: [email protected]; [email protected] 0030-9923/2018/0004-1361 $ 9.00/0 Copyright 2018 Zoological Society of Pakistan Supplementary Material S1.- Primers of the bitter taste receptor genes Tas2r in the raccoon dog. Gene Primer 5’ to 3’ Gene Primer 5' to 3' Tas2r2 CAFA-Tas2r2-R TTTCTCAGGGCTGTATGT Tas2r19 CAFA-Tas2r19P-F CTCCCTCTGCCTATGTCT CAFA-Tas2r2-F AGAAGGAAATTGCCCAAA CAFA-Tas2r19P-R GCAAAGACTGTCCAGGTAA Tas2r5-1 CAFA-Tas2r5-F1 GAGATGAAGACAACGAGGAG Tas2r38 CAFA-Tas2r38-R GGAGGCATATTCGTGAAG CAFA-Tas2r5-R1 ACTTTCCTCAGCGTCTTCTA CAFA-Tas2r38-F ACTGACCCTTCTTTGCTG Tas2r5-2 CAFA-Tas2R5-R2 AGTATAGGCTCAGAGTGC Tas2r39 CAFA-Tas2r39-R GATTGTACCCATGAAGCA CAFA-Tas2R5-F2 GAGATGAAGACAACGAGGAG CAFA-Tas2r39-F TCTCCCTCCAGACACCAA Tas2r7 CAFA-Tas2r7-R GTGCCTATCAAACTACATC Tas2r40 CAFA-Tas2r40-R TGGGTTCCAGTCACAGAG CAFA-Tas2r7-F TAATTCTGAAGGGAGGGA CAFA-Tas2r40-F ATAACATTGAGTGAGGGTC Tas2r8 CAFA-Tas2r8-R TTGCCGGAGAAATGACTA Tas2r41 CAFA-Tas2r41-R AGACATCGGTCTTCTCACATC CAFA-Tas2r8-F GCACTAAACCGCTGGGAC CAFA-Tas2r41-F CCCCATTTACCAGTGTCCC Tas2r9 CAFA-Tas2r9-R CTGGTCCAACAGTGAGGT Tas2r42 CAFA-Tas2r42-R TGTACCCAACAACAGGAG CAFA-Tas2r9-F GCACTAAACCGCTGGGAC CAFA-Tas2r42-F AAAGGAATCCAGCAGAAA Tas2r10 CAFA-Tas2r10-R GCCAACCTTTGTCTCATT Tas2r44 CAFA-Tas2r44-R GTGACGGTGTATTTCTTA CAFA-Tas2r10-F CTGATTGAGCCACCGAGT CAFA-Tas2r44-F CCTGCTTGACATTGGTTA Tas2r12 CAFA-Tas2r12-R GTGTCAGCAGCGGGTAAT Tas2r67 CAFA-Tas2r67-R CTTTCGGGATTCTTTCTA CAFA-Tas2r12-F TTCCCAAGTCTATGTTCGTGT CAFA-Tas2r67-F AGCGAAAGGCTTCACCAT Supplementary Material S2.- Protein sequences of Tas2r genes used in this study. -
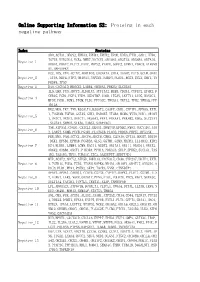
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2