C Onjugate Addition Reactions of Some
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1,1,1,2-Tetrafluoroethane
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Concise International Chemical Assessment Document 11 1,1,1,2-Tetrafluoroethane First draft prepared by Mrs P. Barker and Mr R. Cary, Health and Safety Executive, Liverpool, United Kingdom, and Dr S. Dobson, Institute of Terrestrial Ecology, Huntingdon, United Kingdom Please not that the layout and pagination of this pdf file are not identical to the printed CICAD Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 1998 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organisation (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization -

Functionalization of Heterocycles: a Metal Catalyzed Approach Via
FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION A Dissertation Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Sandeepreddy Vemula In Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Department: Chemistry and Biochemistry October 2018 Fargo, North Dakota North Dakota State University Graduate School Title FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION By Sandeepreddy Vemula The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of DOCTOR OF PHILOSOPHY SUPERVISORY COMMITTEE: Prof. Gregory R. Cook Chair Prof. Mukund P. Sibi Prof. Pinjing Zhao Prof. Dean Webster Approved: November 16, 2018 Prof. Gregory R. Cook Date Department Chair ABSTRACT The central core of many biologically active natural products and pharmaceuticals contain N-heterocycles, the installation of simple/complex functional groups using C-H/N-H functionalization methodologies has the potential to dramatically increase the efficiency of synthesis with respect to resources, time and overall steps to key intermediate/products. Transition metal-catalyzed functionalization of N-heterocycles proved as a powerful tool for the construction of C-C and C-heteroatom bonds. The work in this dissertation describes the development of palladium catalyzed allylation, and the transition metal catalyzed C-H activation for selective functionalization of electron deficient N-heterocycles. Chapter 1 A thorough study highlighting the important developments made in transition metal catalyzed approaches for C-C and C-X bond forming reactions is discussed with a focus on allylation, directed indole C-2 substitution and vinylic C-H activation. -

The Application of the Iodine - Azide Reaction in Thin-Layer Chromatography Studies of Pesticide Preparations
THE APPLICATION OF THE IODINE - AZIDE REACTION IN THIN-LAYER CHROMATOGRAPHY STUDIES OF PESTICIDE PREPARATIONS By T. CSEHHATI* and F.OHSI Department of Biochemistry and Food Chemistry, Technical University Budapest Received October 3, 1981 Presented by Prof. Dr. R. LASZTITY Introduction With chemicals bcing uscd in wider and wider areas for plant protection, toxicologieal and ecological aspects become increasingly important. The formulation analysis of pesticides cannot be limitcd nowadays to the selective determination of the active agent: the identification and quantitative determi nation of impurities is becoming equally significant. The process is of particular importance with phosphoric ester derivatives, since they are more ore less toxic to human beings. Gas chromatographic analysis (GC) of these active agents has been developed many years ago [1]. However, the technique could not supplant thin-layer chromatography (TLC) [2] for the following reasons: a) thermally unstable phosphoric ester derivatives cannot be analyzed by GC; b) if flame ionization detectors are used, one cannot state whether the peaks indicating impurities correspond to presumably toxic side products of the active agents, or to othcr, non-toxic components of the formulation; c) a substantial part of the decomposition products formed in hydrolysis or oxidation proccsses are non-volatile. Consequently, GC methods can only be used when combined with additional processes to yield volatile products; d) the TLC technique, using non-destructive, selective detection processes, allows to isolate, in a much simpler manner as ~ompared to traditional GC, volatile and non-volatile organic phosphoric ester derivatives in amounts in the order of milligrams for further studies of structure identification and toxic properties. -

Used at Rocky Flats
. TASK 1 REPORT (Rl) IDENTIFICATION OF CHEMICALS AND RADIONUCLIDES USED AT ROCKY FLATS I PROJECT BACKGROUND ChemRisk is conducting a Rocky Flats Toxicologic Review and Dose Reconstruction study for The Colorado Department of Health. The two year study will be completed by the fall of 1992. The ChemRisk study is composed of twelve tasks that represent the first phase of an independent investigation of off-site health risks associated with the operation of the Rocky Flats nuclear weapons plant northwest of Denver. The first eight tasks address the collection of historic information on operations and releases and a detailed dose reconstruction analysis. Tasks 9 through 12 address the compilation of information and communication of the results of the study. Task 1 will involve the creation of an inventory of chemicals and radionuclides that have been present at Rocky Flats. Using this inventory, chemicals and radionuclides of concern will be selected under Task 2, based on such factors as the relative toxicity of the materials, quantities used, how the materials might have been released into the environment, and the likelihood for transport of the materials off-site. An historical activities profile of the plant will be constructed under Task 3. Tasks 4, 5, and 6 will address the identification of where in the facility activities took place, how much of the materials of concern were released to the environment, and where these materials went after the releases. Task 7 addresses historic land-use in the vicinity of the plant and the location of off-site populations potentially affected by releases from Rocky Flats. -

Prepared by 3M
DRAFT ECOTOXICOLOGY AND ENVIRONMENTAL FATE TESTING OF SHORT CHAIN PERFLUOROALKYL COMPOUNDS RELATED TO 3M CHEMISTRIES XXX XX, 2008 Prepared by 3M Page 1 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537089 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0001 DRAFT TABLE OF CONTENTS Page Introduction (describes propose, goals, document organization .... ) 3 1) Degradation modes 4-27 Basic degradation pathways, Conversion routes from polymer to monomer & degradants or residual intermediates to degradants) 2) Degradants & Intermediates - Phys. Chem properties and test data by section & chapter i) Fluorinated Sulfonic acids and derivatives a) PFBS (PBSK) 28-42 b) PFBSI 43-44 ii) Fluorinated Carboxylates a) TFA 46-55 b) PFPA 56-59 c) PFBA (APFB) 60-65 d) MeFBSAA (MeFBSE Acid, M370) 66-68 iii) Fluorinated Non-ionics a) MeFBSE 70-74 b) MeFBSA 75-80 c) FBSE 81-84 d) FBSA 85-88 e) HxFBSA 89-91 iv) Fluorinated Inerts & volatiles a) PBSF 94-97 b) NFB, C4 hydride, CF3CF2CF2CF2-H 98-99 3) Assessments X Glossary 100-102 References/Endnotes 105- Page 2 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537090 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0002 DRAFT Introduction to Environmental White Papers on Perfluoroalkyl acids related to 3M chemistries Perfluorochemicals have been commonplace in chemical industry over 50 years but until recently there has been little information on environmental fate and effects avialble in open literature. The following chapters summarize the findings of"list specific C4 intermediates PFBS, PFBSI, PFBA, PFPA, MeFBSAA, TFA, MeFBSE, FBSA, HxFBSA, FBSE, PBSF, NFB " As background, 3M announced on May 16, 2000 the voluntary manufacturing phase out of perfluorooctanyl chemicals which included perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), and PFOS-related chemistries. -

Potentially Explosive Chemicals*
Potentially Explosive Chemicals* Chemical Name CAS # Not 1,1’-Diazoaminonaphthalene Assigned 1,1-Dinitroethane 000600-40-8 1,2,4-Butanetriol trinitrate 006659-60-5 1,2-Diazidoethane 000629-13-0 1,3,5-trimethyl-2,4,6-trinitrobenzene 000602-96-0 1,3-Diazopropane 005239-06-5 Not 1,3-Dinitro-4,5-dinitrosobenzene Assigned Not 1,3-dinitro-5,5-dimethyl hydantoin Assigned Not 1,4-Dinitro-1,1,4,4-tetramethylolbutanetetranitrate Assigned Not 1,7-Octadiene-3,5-Diyne-1,8-Dimethoxy-9-Octadecynoic acid Assigned 1,8 –dihydroxy 2,4,5,7-tetranitroanthraquinone 000517-92-0 Not 1,9-Dinitroxy pentamethylene-2,4,6,8-tetramine Assigned 1-Bromo-3-nitrobenzene 000585-79-5 Not 2,2',4,4',6,6'-Hexanitro-3,3'-dihydroxyazobenzene Assigned 2,2-di-(4,4,-di-tert-butylperoxycyclohexyl)propane 001705-60-8 2,2-Dinitrostilbene 006275-02-1 2,3,4,6- tetranitrophenol 000641-16-7 Not 2,3,4,6-tetranitrophenyl methyl nitramine Assigned Not 2,3,4,6-tetranitrophenyl nitramine Assigned Not 2,3,5,6- tetranitroso nitrobenzene Assigned Not 2,3,5,6- tetranitroso-1,4-dinitrobenzene Assigned 2,4,6-Trinitro-1,3,5-triazo benzene 029306-57-8 Not 2,4,6-trinitro-1,3-diazabenzene Assigned Not 2,4,6-Trinitrophenyl trimethylol methyl nitramine trinitrate Assigned Not 2,4,6-Trinitroso-3-methyl nitraminoanisole Assigned 2,4-Dinitro-1,3,5-trimethyl-benzene 000608-50-4 2,4-Dinitrophenylhydrazine 000119-26-6 2,4-Dinitroresorcinol 000519-44-8 2,5-dimethyl-2,5-diydroperoxy hexane 2-Nitro-2-methylpropanol nitrate 024884-69-3 3,5-Dinitrosalicylic acid 000609-99-4 Not 3-Azido-1,2-propylene glycol dinitrate -

Synthesis with Iodine Azide
Controlling hazardous chemicals in microreactors: Synthesis with iodine azide Johan C. Brandt and Thomas Wirth* Full Research Paper Open Access Address: Beilstein Journal of Organic Chemistry 2009, 5, No. 30. Cardiff University, School of Chemistry, Park Place, Cardiff CF10 doi:10.3762/bjoc.5.30 3AT, UK. Received: 23 March 2009 Email: Accepted: 04 June 2009 Thomas Wirth* - [email protected] Published: 12 June 2009 * Corresponding author Guest Editor: A. Kirschning Keywords: © 2009 Brandt and Wirth; licensee Beilstein-Institut. azide; flow chemistry; hazardous reagents; microreactor; License and terms: see end of document. rearrangement Abstract Aromatic aldehydes have been converted into the corresponding carbamoyl azides using iodine azide. These reactions have been performed safely under continuous flow reaction conditions in microreactors. Introduction Microstructured devices have already found their way into moieties in organic synthesis as they can be transformed easily organic synthesis, because they offer various advantages over into a large variety of other functional groups [5]. traditional large-scale chemistry performed in flasks or vessels [1,2]. The high surface-to-volume ratio as their main character- Iodine azide is a very hazardous but valuable reagent that is istic can result in rapid mixing of compounds, uniform reaction easier and much more safely handled under microreactor condi- conditions due to precise temperature control as well as rate tions. Iodine azide is a solid compound, highly explosive and enhancements because of short diffusion distances. In addition, toxic. It is known to add stereospecifically to carbon–carbon the synthesis and use of potentially hazardous compounds is double bonds with high regioselectivity following an ionic another advantage of microreactors as only very small amounts mechanism. -

1.Brethericks1 51 1..51
0001. Silver [7440-22-4] Ag Ag Acetylenic compounds MRH Acetylene 8.70/99þ See ACETYLENIC COMPOUNDS Aziridine See Aziridine: Silver Bromine azide See Bromine azide 3-Bromopropyne See 3-Bromopropyne: Metals Carboxylic acids Koffolt, J. H., private comm., 1965 Silver is incompatible with oxalic or tartaric acids, since the silver salts decompose on heating. Silver oxalate explodes at 140C, and silver tartrate loses carbon dioxide. See other METAL OXALATES Chlorine trifluoride MRH 1.42/36 See Chlorine trifluoride: Metals Copper, Ethylene glycol See Ethylene glycol: Silvered copper wire Electrolytes, Zinc Britz, W. K. et al., Chem. Abs., 1975, 83, 150293 Causes of spontaneous combustion and other hazards of silver—zinc batteries were investigated. Ethanol, Nitric acid Luchs, J. K., Photog. Sci. Eng., 1966, 10, 334 Action of silver on nitric acid in presence of ethanol may form the readily detonable silver fulminate. See Nitric acid: Alcohols See also SILVER-CONTAINING EXPLOSIVES Ethyl hydroperoxide See Ethyl hydroperoxide: Silver Ethylene oxide MRH 3.72/99þ See Ethylene oxide: Reference 4 Hydrogen peroxide MRH 1.59/99þ See Hydrogen peroxide: Metals Iodoform Grignard, 1935, Vol. 3, 320 In contact with finely divided (reduced) silver, incandescence occurs. 1 Other reactants Yoshida, 1980, 103 MRH values for 7 combinations, largely with catalytically susceptible materials, are given. Ozonides See OZONIDES Peroxomonosulfuric acid See Peroxomonosulfuric acid: Catalysts Peroxyformic acid MRH 5.69/100 See Peroxyformic acid: Metals See other METALS 0002. Silver—aluminium alloy [11144-29-9] AgÀAl Ag Al 1. Popov, E. I. et al., Chem. Abs., 1977, 87, 205143 2. Popov, E. I. et al., Chem. -

Poly(Limonene Carbonate): a Bio-Based & Versatile High-Performance Thermoplastic
Poly(limonene carbonate): a bio-based & versatile high-performance thermoplastic Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. Nat.) an der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth vorgelegt von Oliver Hauenstein aus Dortmund Bayreuth 2016 Die vorliegende Arbeit wurde in der Zeit von November 2012 bis Juni 2016 in Bayreuth am Lehrstuhl Makromolekulare Chemie II unter Betreuung von Herrn Professor Dr. Andreas Greiner angefertigt. Vollständiger Abdruck der von der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth genehmigten Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.). Dissertation eingereicht am: 13.07.2016 Zulassung durch das Leitungsgremium: 26.07.2016 Wissenschaftliches Kolloquium: 01.02.2017 Amtierender Direktor: Prof. Dr. Stephan Kümmel Prüfungsausschuss: Prof. Dr. Andreas Greiner (Erstgutachter) Prof. Dr. Hans-Werner Schmidt (Zweitgutachter) Prof. Dr. Carlo Unverzagt. (Vorsitz) Prof. Dr. Jürgen Senker Drittgutachter: Prof. Dr. Volker Abetz Für meine Familie & Hui Contents Abbreviations & symbols ......................................................................................................................... i List of publications ................................................................................................................................... v Abstract .................................................................................................................................................... -

Multistep Flow Synthesis of Vinyl Azides and Their Use in the Copper-Catalyzed Huisgen-Type Cycloaddition Under Inductive-Heating Conditions
Multistep flow synthesis of vinyl azides and their use in the copper-catalyzed Huisgen-type cycloaddition under inductive-heating conditions Lukas Kupracz, Jan Hartwig, Jens Wegner, Sascha Ceylan and Andreas Kirschning* Full Research Paper Open Access Address: Beilstein J. Org. Chem. 2011, 7, 1441–1448. Institute of Organic Chemistry, Leibniz University Hannover, doi:10.3762/bjoc.7.168 Schneiderberg 1b, 30167 Hannover, Germany Received: 21 August 2011 Email: Accepted: 07 October 2011 Lukas Kupracz - [email protected]; Jan Hartwig - Published: 20 October 2011 [email protected]; Jens Wegner - [email protected]; Andreas Kirschning* - This article is part of the Thematic Series "Chemistry in flow systems II". [email protected] Associate Editor: J. Murphy * Corresponding author © 2011 Kupracz et al; licensee Beilstein-Institut. Keywords: License and terms: see end of document. flow reactor; inductive heating; iodine azide; polymer-supported reagents; vinyl azides Abstract The multistep flow synthesis of vinyl azides and their application in the synthesis of vinyltriazoles is reported. The synthesis relies on a stable polymer-bound equivalent of iodine azide that serves to carry out 1,2-functionalization of alkenes in a telescope flow protocol. The intermediate 2-iodo azides are subjected to a DBU-mediated polymer-supported elimination step yielding vinyl azides in good yield. The third step involves the formation of vinyl triazoles by a copper-catalyzed Huisgen-"click" cycloaddition. The required heat is generated by electromagnetic induction based on copper. Copper serves both as heatable as well as catalytically active packed-bed material inside the flow reactor. -
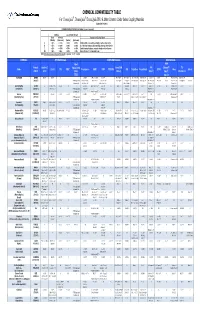
CPC Chemical Compatibility Chart
CHEMICAL COMPATIBILITY TABLE For ChemQuik®, DrumQuik®, DrumQuik PRO & Other Common Colder Series Coupling Materials (Updated 01/14/2010) INTERPRETATION OF TEST DATA (In 30 days to 1 year of exposure) Swelling Loss of Tensile Strength Linear Volumetric Description of Chemical Attack (Plastics) (Elastomers) (Plastics) (Elastomers) A < 10% <= 15% < 15% <=15% Excellent, little or no swelling, softening or surface deterioration B < 15% <= 30% < 30% <= 30% Good chemical resistance, minor swelling, softening or deterioration C < 20% <= 50% < 50% <= 60% Limited chemical resistance, moderate attack, conditional service NR > 20% > 50% > 50% > 60% Severe attack, not recommended for use NOTE: All temperatures are in degrees Fahrenheit. Conversion: °C = (°F - 32)/1.8 CHEMICAL SPRING Materials COUPLING Materials SEAL Materials Teflon® FFKM ® Formula Hastelloy C Encapsulated Acetal/POM FKM (Chemraz / TPO Name 316 SS PPS PEEK™ Polypropylene HDPE PVDF PTFE/PFA ABS Polysulfone Polycarbonate EPDM Buna Silicone (CAS #) (276) 316SS (Celcon) (Viton®) Simriz® / (Santoprene) (TESS) Kalrez®) Acetic Acid C2H4O2 A to 212° 212° A to 212° 212° A A A A to 140° 140° AB to 100% to 70° 70° A to 122° 122° A AAto5%to70 to 5% to 70°° AB 10% to 70° 70° A to 100% to 70° 70° A to 50% to 70° 70° A 10% to 70° 70° AAto70 to 70°° A B to 30% at 70° 70° A to 30% to 70° 70° A (64-19-7) (PTFE Encapsulated AB 50-100% to 160° AB 60% to 180° A to 10% to 225° BC 10% @ 70° C 20% @ 70° A to 20% to 140° B to 50% @ 122° B 10-25% to 100° AB to 200° A to 70° B to 20% to 185° C 50% @ 70° A to -

1,1,1,2-Tetrafluoroethane (HFC-134A) (CAS No. 811-97-2) (Second Edition)
1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) JACC No. 50 ISSN-0773-6339-50 Brussels, January 2006 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) ECETOC JACC REPORT No. 50 © Copyright – ECETOC AISBL European Centre for Ecotoxicology and Toxicology of Chemicals 4 Avenue E. Van Nieuwenhuyse (Bte 6), B-1160 Brussels, Belgium. All rights reserved. No part of this publication may be reproduced, copied, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the copyright holder. Applications to reproduce, store, copy or translate should be made to the Secretary General. ECETOC welcomes such applications. Reference to the document, its title and summary may be copied or abstracted in data retrieval systems without subsequent reference. The content of this document has been prepared and reviewed by experts on behalf of ECETOC with all possible care and from the available scientific information. It is provided for information only. ECETOC cannot accept any responsibility or liability and does not provide a warranty for any use or interpretation of the material contained in the publication. ECETOC JACC No. 50 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) CONTENTS EXECUTIVE SUMMARY 1 THE ECETOC SCHEME FOR THE JOINT ASSESSMENT OF COMMODITY CHEMICALS 3 1. SUMMARY AND CONCLUSIONS 4 2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS 6 2.1 Identity 6 2.2 EU classification and labelling 6 2.3 Physical and chemical properties 6 2.4 Conversion factors 8 2.5 Analytical methods 8 3.