Synthetic and Structural Studies on Natural Coumarins, THESIS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1,1,1,2-Tetrafluoroethane
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Concise International Chemical Assessment Document 11 1,1,1,2-Tetrafluoroethane First draft prepared by Mrs P. Barker and Mr R. Cary, Health and Safety Executive, Liverpool, United Kingdom, and Dr S. Dobson, Institute of Terrestrial Ecology, Huntingdon, United Kingdom Please not that the layout and pagination of this pdf file are not identical to the printed CICAD Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 1998 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organisation (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization -

Functionalization of Heterocycles: a Metal Catalyzed Approach Via
FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION A Dissertation Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Sandeepreddy Vemula In Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Department: Chemistry and Biochemistry October 2018 Fargo, North Dakota North Dakota State University Graduate School Title FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION By Sandeepreddy Vemula The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of DOCTOR OF PHILOSOPHY SUPERVISORY COMMITTEE: Prof. Gregory R. Cook Chair Prof. Mukund P. Sibi Prof. Pinjing Zhao Prof. Dean Webster Approved: November 16, 2018 Prof. Gregory R. Cook Date Department Chair ABSTRACT The central core of many biologically active natural products and pharmaceuticals contain N-heterocycles, the installation of simple/complex functional groups using C-H/N-H functionalization methodologies has the potential to dramatically increase the efficiency of synthesis with respect to resources, time and overall steps to key intermediate/products. Transition metal-catalyzed functionalization of N-heterocycles proved as a powerful tool for the construction of C-C and C-heteroatom bonds. The work in this dissertation describes the development of palladium catalyzed allylation, and the transition metal catalyzed C-H activation for selective functionalization of electron deficient N-heterocycles. Chapter 1 A thorough study highlighting the important developments made in transition metal catalyzed approaches for C-C and C-X bond forming reactions is discussed with a focus on allylation, directed indole C-2 substitution and vinylic C-H activation. -

Prepared by 3M
DRAFT ECOTOXICOLOGY AND ENVIRONMENTAL FATE TESTING OF SHORT CHAIN PERFLUOROALKYL COMPOUNDS RELATED TO 3M CHEMISTRIES XXX XX, 2008 Prepared by 3M Page 1 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537089 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0001 DRAFT TABLE OF CONTENTS Page Introduction (describes propose, goals, document organization .... ) 3 1) Degradation modes 4-27 Basic degradation pathways, Conversion routes from polymer to monomer & degradants or residual intermediates to degradants) 2) Degradants & Intermediates - Phys. Chem properties and test data by section & chapter i) Fluorinated Sulfonic acids and derivatives a) PFBS (PBSK) 28-42 b) PFBSI 43-44 ii) Fluorinated Carboxylates a) TFA 46-55 b) PFPA 56-59 c) PFBA (APFB) 60-65 d) MeFBSAA (MeFBSE Acid, M370) 66-68 iii) Fluorinated Non-ionics a) MeFBSE 70-74 b) MeFBSA 75-80 c) FBSE 81-84 d) FBSA 85-88 e) HxFBSA 89-91 iv) Fluorinated Inerts & volatiles a) PBSF 94-97 b) NFB, C4 hydride, CF3CF2CF2CF2-H 98-99 3) Assessments X Glossary 100-102 References/Endnotes 105- Page 2 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537090 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0002 DRAFT Introduction to Environmental White Papers on Perfluoroalkyl acids related to 3M chemistries Perfluorochemicals have been commonplace in chemical industry over 50 years but until recently there has been little information on environmental fate and effects avialble in open literature. The following chapters summarize the findings of"list specific C4 intermediates PFBS, PFBSI, PFBA, PFPA, MeFBSAA, TFA, MeFBSE, FBSA, HxFBSA, FBSE, PBSF, NFB " As background, 3M announced on May 16, 2000 the voluntary manufacturing phase out of perfluorooctanyl chemicals which included perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), and PFOS-related chemistries. -

Antiproliferative Effect of Angelica Archangelica Fruits Steinthor Sigurdssona,*, Helga M
Antiproliferative Effect of Angelica archangelica Fruits Steinthor Sigurdssona,*, Helga M. Ögmundsdottirb, and Sigmundur Gudbjarnasona a Science Institute, University of Iceland, Vatnsmyrarvegur 16, IS-101 Reykjavik, Iceland. Fax: +354 525 4886. E-mail: [email protected] b Molecular and Cell Biology Research Laboratory, Icelandic Cancer Society, Skogarhlid 8, IS-101 Reykjavik, Iceland * Author for correspondence and reprint requests Z. Naturforsch. 59c, 523Ð527 (2004); received December 21, 2003/February 9, 2004 The aim of this work was to study the antiproliferative effect of a tincture from fruits of Angelica archangelica and the active components using the human pancreas cancer cell line PANC-1 as a model. Significant dose-dependent antiproliferative activity was observed in µ the tincture with an EC50 value of 28.6 g/ml. Strong antiproliferative activity resulted from the two most abundant furanocoumarins in the tincture, imperatorin and xanthotoxin. The contribution of terpenes to this activity was insignificant. Imperatorin and xanthotoxin µ µ proved to be highly antiproliferative, with EC50 values of 2.7 g/ml and 3.7 g/ml, respec- tively, equivalent to 10 and 17 µm. The results indicate that furanocoumarins account for most of the antiproliferative activity of the tincture. Key words: Angelica archangelica, Xanthotoxin, Imperatorin Introduction their inhibiting effect on cytochrome P450, result- Angelica archangelica has been long and widely ing in drug-interactions (Guo et al., 2000; Koenigs used in folk medicine, and it is one of the most and Trager, 1998; Zhang et al., 2001). Imperatorin respected medicinal herbs in Nordic countries, has also been found to decrease chemically in- where it was cultivated during the Middle Ages, duced DNA adduct formation and may thus pos- and exported to other parts of Europe. -

Poly(Limonene Carbonate): a Bio-Based & Versatile High-Performance Thermoplastic
Poly(limonene carbonate): a bio-based & versatile high-performance thermoplastic Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. Nat.) an der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth vorgelegt von Oliver Hauenstein aus Dortmund Bayreuth 2016 Die vorliegende Arbeit wurde in der Zeit von November 2012 bis Juni 2016 in Bayreuth am Lehrstuhl Makromolekulare Chemie II unter Betreuung von Herrn Professor Dr. Andreas Greiner angefertigt. Vollständiger Abdruck der von der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth genehmigten Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.). Dissertation eingereicht am: 13.07.2016 Zulassung durch das Leitungsgremium: 26.07.2016 Wissenschaftliches Kolloquium: 01.02.2017 Amtierender Direktor: Prof. Dr. Stephan Kümmel Prüfungsausschuss: Prof. Dr. Andreas Greiner (Erstgutachter) Prof. Dr. Hans-Werner Schmidt (Zweitgutachter) Prof. Dr. Carlo Unverzagt. (Vorsitz) Prof. Dr. Jürgen Senker Drittgutachter: Prof. Dr. Volker Abetz Für meine Familie & Hui Contents Abbreviations & symbols ......................................................................................................................... i List of publications ................................................................................................................................... v Abstract .................................................................................................................................................... -
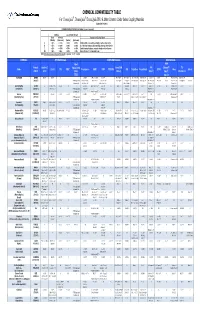
CPC Chemical Compatibility Chart
CHEMICAL COMPATIBILITY TABLE For ChemQuik®, DrumQuik®, DrumQuik PRO & Other Common Colder Series Coupling Materials (Updated 01/14/2010) INTERPRETATION OF TEST DATA (In 30 days to 1 year of exposure) Swelling Loss of Tensile Strength Linear Volumetric Description of Chemical Attack (Plastics) (Elastomers) (Plastics) (Elastomers) A < 10% <= 15% < 15% <=15% Excellent, little or no swelling, softening or surface deterioration B < 15% <= 30% < 30% <= 30% Good chemical resistance, minor swelling, softening or deterioration C < 20% <= 50% < 50% <= 60% Limited chemical resistance, moderate attack, conditional service NR > 20% > 50% > 50% > 60% Severe attack, not recommended for use NOTE: All temperatures are in degrees Fahrenheit. Conversion: °C = (°F - 32)/1.8 CHEMICAL SPRING Materials COUPLING Materials SEAL Materials Teflon® FFKM ® Formula Hastelloy C Encapsulated Acetal/POM FKM (Chemraz / TPO Name 316 SS PPS PEEK™ Polypropylene HDPE PVDF PTFE/PFA ABS Polysulfone Polycarbonate EPDM Buna Silicone (CAS #) (276) 316SS (Celcon) (Viton®) Simriz® / (Santoprene) (TESS) Kalrez®) Acetic Acid C2H4O2 A to 212° 212° A to 212° 212° A A A A to 140° 140° AB to 100% to 70° 70° A to 122° 122° A AAto5%to70 to 5% to 70°° AB 10% to 70° 70° A to 100% to 70° 70° A to 50% to 70° 70° A 10% to 70° 70° AAto70 to 70°° A B to 30% at 70° 70° A to 30% to 70° 70° A (64-19-7) (PTFE Encapsulated AB 50-100% to 160° AB 60% to 180° A to 10% to 225° BC 10% @ 70° C 20% @ 70° A to 20% to 140° B to 50% @ 122° B 10-25% to 100° AB to 200° A to 70° B to 20% to 185° C 50% @ 70° A to -

1,1,1,2-Tetrafluoroethane (HFC-134A) (CAS No. 811-97-2) (Second Edition)
1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) JACC No. 50 ISSN-0773-6339-50 Brussels, January 2006 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) ECETOC JACC REPORT No. 50 © Copyright – ECETOC AISBL European Centre for Ecotoxicology and Toxicology of Chemicals 4 Avenue E. Van Nieuwenhuyse (Bte 6), B-1160 Brussels, Belgium. All rights reserved. No part of this publication may be reproduced, copied, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the copyright holder. Applications to reproduce, store, copy or translate should be made to the Secretary General. ECETOC welcomes such applications. Reference to the document, its title and summary may be copied or abstracted in data retrieval systems without subsequent reference. The content of this document has been prepared and reviewed by experts on behalf of ECETOC with all possible care and from the available scientific information. It is provided for information only. ECETOC cannot accept any responsibility or liability and does not provide a warranty for any use or interpretation of the material contained in the publication. ECETOC JACC No. 50 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) CONTENTS EXECUTIVE SUMMARY 1 THE ECETOC SCHEME FOR THE JOINT ASSESSMENT OF COMMODITY CHEMICALS 3 1. SUMMARY AND CONCLUSIONS 4 2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS 6 2.1 Identity 6 2.2 EU classification and labelling 6 2.3 Physical and chemical properties 6 2.4 Conversion factors 8 2.5 Analytical methods 8 3. -

Ethyl Acetate Extract Components of Bushen-Yizhi Formula Provides
www.nature.com/scientificreports Corrected: Author Correction OPEN Ethyl Acetate Extract Components of Bushen-Yizhi Formula Provides Neuroprotection against Received: 9 January 2017 Accepted: 9 August 2017 Scopolamine-induced Cognitive Published online: 29 August 2017 Impairment Shi-Jie Zhang1, Dan Luo1, Lin Li1, Rui-Rong Tan2, Qing-Qing Xu1, Jie Qin3, Lei Zhu1, Na-Chuan Luo1, Ting-Ting Xu1, Rong Zhang1, Lei Yang1 & Qi Wang1 Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder and there is no efective cure for this devastating disease to date. Bushen Yizhi Formula (BSYZ-F), a Chinese herbal compound, has proved to be efective for AD. In this study, we further investigate the efective part of BSYZ-F, ethyl acetate extract components of BSYZ-F (BSYZ-E), protects scopolamine (SCOP)-induced cognitive impairment, which shows a similar efect to BSYZ-F. We also fnd that BSYZ-E could protect against SCOP-induced cholinergic system dysfunction. In neuron function level, BSYZ-E remarkably elevates protein levels of nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). BSYZ-E also signifcantly mitigates SCOP-induced apoptosis, oxidative stress and nitrosative stress. Conclusively, BSYZ-E, the efective part of BSYZ-F, can provide neuroprotection against SCOP-induced cognitive impairment through a multifunctional strategy. These fndings suggest that BSYZ-E might be developed as a therapeutic drug for AD by targeting multiple pathways of the pathogenesis. Alzheimer’s disease (AD) is a chronic neurodegenerative brain disorder. AD is characterized by progressive loss of neurons mainly in hippocampus and cortex, which results in dysfunctions of cognition and emotion1. AD afects people aged 65 and older most commonly2, 3. -

Opinion of the Sccnfp on Isopimpinellin
SCCNFP/0761/03 OPINION OF THE SCIENTIFIC COMMITTEE ON COSMETIC PRODUCTS AND NON-FOOD PRODUCTS INTENDED FOR CONSUMERS CONCERNING ISOPIMPINELLIN adopted by the SCCNFP during the 26th plenary meeting of of 9 December 2003 1. Terms of Reference SCCNFP/0761/03 Evaluation and opinion on Isopimpinellin ____________________________________________________________________________________________ 1.1 Context of the question The adaptation to technical progress of the Annexes to Council Directive 76/768/EEC of 27 July 1976 on the approximation of the laws of the Member States relating to cosmetic products. 1.2 Request to the SCCNFP The SCCNFP is requested to answer the following question : • Does the data provided justify an update of the “Initial List of fragrance” for No 6 of the table attached to this opinion (An Initial List of Perfumery Materials which must not form part of Cosmetic Products except subject to the restrictions and conditions laid down [SCCNFP/0392, Adopted 25.09.01]) and how should the restrictions and conditions laid down be changed accordingly? The restriction in No 6 reads: May be used in cosmetic products, provided that the total concentration of furocumarin-like substances in the finished cosmetic product do not exceed 1 ppm. The present Opinion will primarily deal with questions concerning photomutagenicity of isopimpinellin. 1.3 Statement on the toxicological evaluation The SCCNFP is the scientific advisory body to the European Commission in matters of consumer protection with respect to cosmetics and non-food products intended for consumers. The Commission’s general policy regarding research on animals supports the development of alternative methods to replace or to reduce animal testing when possible. -

Snider, Barry B
BARRY B. SNIDER Dept of Chemistry, MS 015 Brandeis University Waltham, MA 02454-9110 Born: 1950, Chicago Illinois Education: B. Sc. Chem. University of Michigan, Ann Arbor, MI 1970 Ph. D. Harvard University, Cambridge, MA 1973 (Advisor, E. J. Corey) Postdoctoral Columbia University, New York, NY 1973-1975 (Advisor, Ronald Breslow) Employment Assistant Professor, Princeton University, 1975-1981 Associate Professor, Brandeis University, 1981-1985 Professor, Brandeis University, 1985- Breskin Professor of Organic Chemistry, Brandeis University, 1998- Chair, Chemistry Department, Brandeis University, 1992-1995, 2013-present Awards and Honors National Science Foundation Predoctoral Fellowship 1970-1973 National Institutes of Health Postdoctoral Fellowship, 1975 DuPont Young Faculty Grant, 1978, 1979 UpJohn Young Faculty Grant, 1977, 1978 Fellow of the Alfred P. Sloan Foundation 1979-1983 Dreyfus Foundation Teacher-Scholar Award, 1982-1987 Visiting Research Scholar, Tokyo Institute of Technology, 1992 Member, NIH Medicinal Chemistry Study Section, 1992-1997 Chair, NIH Medicinal Chemistry Study Section, 1995-1997 Cope Scholar Award (American Chemical Society), 1995 Japan Society for Promotion of Science Fellowship, 1999 Honorary Professor East China University of Science and Technology 2006 Chair-Elect, Chair, Past Chair, Organic Chemistry Division of the ACS, 2006-2008 Fellow, American Chemical Society, 2011 Secretary/Treasurer, Organic Chemistry Division of the ACS, (Elect 2013) 2014-2017 Memberships: Sigma Xi, Phi Beta Kappa, American Chemical Society Link to Web page and complete publication list: Barry Snider faculty webpage Snider Publication List Page 2/24 Publications 1. Salamone, J. C.; Snider, B. “Quaternary Ammonium Polymers from 1,4- Diaza[2.2.2]bicyclooctane” J. Polym. Sci. A-1 1970, 8, 3495-3501. -

Angelica Archangelica L
Swedish University of Agricultural Sciences The Faculty of landscape Planning, Horticulture and Agriculture Science Plant Breeding and Biotechnology Angelica archangelica L. Madeleine Kylin Degree project • 15 hec • Basic C BSc in Horticulture programme • Självständigt arbete vid LTJ-fakulteten, SLU Alnarp 2010 1 2 Angelica archangelica L. Madeleine Kylin Supervisor: Björn Salomon, Swedish University of Agricultural Sciences (SLU), Plant Breeding and Biotechnology Examiner: Kimmo Rumpunen , Swedish University of Agricultural Sciences (SLU), Department of Plant Breeding and Biotechnology Credits: 15 hec Level: Basic C Course title: Degree Project for BSc Thesis in Horticulture Course code: EX0369 Programme/education: BSc in Horticulture programme Place of publication: Alnarp Year of publication: 2010 Title of series: Självständigt arbete vid LTJ-fakulteten Online publication: http://stud.epsilon.slu.se Key Words: Angelica archangelica L.,subsp. archangelica, subsp. litoralis Swedish University of Agricultural Sciences The Faculty of landscape Planning, Horticulture and Agriculture Science Plant Breeding and Biotechnology 3 4 Abstract Angelica archangelica (Garden angelica) is the only Medicinal and Aromatic Plant (MAP) with a Nordic origin. The plant can reach up to three meters when cultivated. Angelica archangelica is used as flavouring in additives, honey, beverage base, essential oils, fol- klore medicine and as ornamental for decorative purposes. Commercial cultivation is mainly focused on root production. Production countries are Poland, Netherland, France, Belgium, Switzerland, and former Czechoslovakia with an overall yearly world production of 1000 kg of essential oils. Compounds to be found in A.archangelica are bittering agents, essential oils, flavonoids, tanning agents, resins, silica, carbohydrates, coumarins, organic acids and terpenes. Essential oils derived from angelica root are composed of over 60 dif- ferent constituents. -

Tolerance and Metabolism of Furanocoumarins by the Phytopathogenic Fungus Gibberella Pulicaris (Fusarium Sambucinum)
Phytochemistry, Vol. 28, No. II, pp. 2963-2969,1989. 0031-9422/89 S3.00 +0.00 Printed in Great Britain. © 1989 Pergamon Press pic TOLERANCE AND METABOLISM OF FURANOCOUMARINS BY THE PHYTOPATHOGENIC FUNGUS GIBBERELLA PULICARIS (FUSARIUM SAMBUCINUM) ANNE E. DESJARDINS,* GAYLAND F. SPENCER and RONALD D. PLATTNER U.S. Department of Agriculture, Agricultural Research Service, Northern Regional Research Center, 1815 North University Street, Peoria, IL 61604, U.S.A. (Received in revised form 3 April 1989) Key Word Index-Pastinaca sativa; Umbellifereae; parsnip; Gibberella pulicaris (Fusarium sambucinum); phyto alexin metabolism; furanocoumarins. Abstract-Sixty-two strains of Gibberella pulicaris (anamorph: Fusarium sambucinum) from diseased plants and from soil were tested for tolerance of the furanocoumarin xanthotoxin in vitro. Twenty-one (88%) of the plant-derived strains and two (5%) ofthe soil-derived strains were highly tolerant ofxanthotoxin. Sixteen selected strains were tested further against 16 furanocoumarins or furanocoumarin precursors. All plant-derived strains tested were highly tolerant of and, in most cases, able to completely metabolize all 16 compounds. Most soil-derived strains tested were tolerant of furanocoumarin precursors but sensitive to certain furanocoumarins. Linear compounds methoxylated at C-8 appeared more toxic than both those unsubstituted and those with longer-chain ethers. Tolerance of angelicin, xanthotoxin, pimpinellin and isopimpinellin correlated in large part with their metabolism. All strains that were