Functionalization of Heterocycles: a Metal Catalyzed Approach Via
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1,1,1,2-Tetrafluoroethane
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Concise International Chemical Assessment Document 11 1,1,1,2-Tetrafluoroethane First draft prepared by Mrs P. Barker and Mr R. Cary, Health and Safety Executive, Liverpool, United Kingdom, and Dr S. Dobson, Institute of Terrestrial Ecology, Huntingdon, United Kingdom Please not that the layout and pagination of this pdf file are not identical to the printed CICAD Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 1998 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organisation (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization -

Synthesis of Oxymethylene Ethers
Synthesis of Oxymethylene Ethers Dissertation Zur Erlangung des akademischen Grades Doktor der Naturwissenschaften (Dr. rer. nat.) vorgelegt an der Fakultät für Chemie und Biochemie der Ruhr-Universität Bochum von Anna Grünert Bochum Oktober 2019 Die vorliegende Arbeit wurde in der Zeit von Februar 2016 bis Oktober 2019 in der Abteilung für Heterogene Katalyse am Max-Planck-Institut für Kohlenforschung in Mülheim an der Ruhr unter der Leitung von Prof. Dr. Ferdi Schüth angefertigt. Referent: Prof. Dr. Ferdi Schüth Korreferent: Prof. Dr. Martin Muhler I I Acknowledgements Firstly, I would like to thank my team of supervisors, Prof. Dr. Ferdi Schüth and Dr. Wolfgang Schmidt, and my co-examiner Prof. Dr. Martin Muhler. Ferdi, I would like to thank you for your trust that is the basis of the exceptional freedom of work, which you grant not only to me, but to all of your PhD students. You gave me the resources, time and liberty to develop my PhD project in my own way, to make mistakes, to solve challenging problems and to grow as a person. I am grateful for your appreciation of a cooperative and welcoming atmosphere in the group, which is most apparent in your yearly invitation to the group trip to Oberwesel. Wolfgang, I owe my thanks to you for your advice on many topics including material synthesis, catalyst characterisation and manuscript writing. I very much appreciate your welcoming and relaxed attitude. I am also thankful that to you both that you enjoy sharing your knowledge and experience with me and my colleagues in catalysis seminars and other technical seminars and in the focused project meetings. -

Prepared by 3M
DRAFT ECOTOXICOLOGY AND ENVIRONMENTAL FATE TESTING OF SHORT CHAIN PERFLUOROALKYL COMPOUNDS RELATED TO 3M CHEMISTRIES XXX XX, 2008 Prepared by 3M Page 1 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537089 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0001 DRAFT TABLE OF CONTENTS Page Introduction (describes propose, goals, document organization .... ) 3 1) Degradation modes 4-27 Basic degradation pathways, Conversion routes from polymer to monomer & degradants or residual intermediates to degradants) 2) Degradants & Intermediates - Phys. Chem properties and test data by section & chapter i) Fluorinated Sulfonic acids and derivatives a) PFBS (PBSK) 28-42 b) PFBSI 43-44 ii) Fluorinated Carboxylates a) TFA 46-55 b) PFPA 56-59 c) PFBA (APFB) 60-65 d) MeFBSAA (MeFBSE Acid, M370) 66-68 iii) Fluorinated Non-ionics a) MeFBSE 70-74 b) MeFBSA 75-80 c) FBSE 81-84 d) FBSA 85-88 e) HxFBSA 89-91 iv) Fluorinated Inerts & volatiles a) PBSF 94-97 b) NFB, C4 hydride, CF3CF2CF2CF2-H 98-99 3) Assessments X Glossary 100-102 References/Endnotes 105- Page 2 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537090 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0002 DRAFT Introduction to Environmental White Papers on Perfluoroalkyl acids related to 3M chemistries Perfluorochemicals have been commonplace in chemical industry over 50 years but until recently there has been little information on environmental fate and effects avialble in open literature. The following chapters summarize the findings of"list specific C4 intermediates PFBS, PFBSI, PFBA, PFPA, MeFBSAA, TFA, MeFBSE, FBSA, HxFBSA, FBSE, PBSF, NFB " As background, 3M announced on May 16, 2000 the voluntary manufacturing phase out of perfluorooctanyl chemicals which included perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), and PFOS-related chemistries. -

Novel Microbiocides
(19) TZZ _Z__T (11) EP 2 641 901 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 25.09.2013 Bulletin 2013/39 C07D 215/40 (2006.01) C07D 401/12 (2006.01) A61K 31/4709 (2006.01) A01N 43/42 (2006.01) (21) Application number: 12160780.8 (22) Date of filing: 22.03.2012 (84) Designated Contracting States: (72) Inventor: The designation of the inventor has not AL AT BE BG CH CY CZ DE DK EE ES FI FR GB yet been filed GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (74) Representative: Herrmann, Jörg Designated Extension States: Syngenta Crop Protection BA ME Münchwilen AG Intellectual Property Department (71) Applicant: Syngenta Participations AG Schaffhauserstrasse 4058 Basel (CH) 4332 Stein (CH) (54) Novel microbiocides (57) The invention relates to compounds of formula I wherein R1, R2, X, Y1, Y2, Y3, D1, D2, D3, G1, G2, G3 and p are as defined in the claims. The invention further provides intermediates used in the preparation of these compounds, to compositions which comprise these compounds and to theiruse in agriculture or horticulture for controlling orpreventing infestation of plants by phytopathogenic microorganisms, preferably fungi. EP 2 641 901 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 641 901 A1 Description [0001] The present invention relates to novel microbiocidally active, in particular fungicidally active, cyclic bisoxime derivatives. Itfurther relatesto intermediates used inthe preparationof these compounds, to compositions which comprise 5 these compounds and to their use in agriculture or horticulture for controlling or preventing infestation of plants by phytopathogenic microorganisms, preferably fungi. -

Chemical Innovation Technologies to Make Processes and Products More Sustainable
United States Government Accountability Office Center for Science, Technology, and Engineering Natural Resources and Environment Report to Congressional Requesters February 2018 TECHNOLOGY ASSESSMENT Chemical Innovation Technologies to Make Processes and Products More Sustainable GAO-18-307 The cover image displays a word cloud generated from the transcript of the meeting we convened with 24 experts in the field of sustainable chemistry. The size of the words in the cloud corresponds to the frequency with which each word appeared in the transcript. In most cases, similar words—such as singular and plural versions of the same word— were combined into a single term. Words that were unrelated to the topic of sustainable chemistry were removed. The images around the periphery are stylized representations of chemical molecules that seek to illustrate a new conceptual framework, whereby molecules can be transformed to provide better performance; however, they are not intended to represent specific chemical compounds. TECHNOLOGY ASSESSMENT Highlights of GAO-18-307, a report to congressional requesters Chemical Innovation February 2018 Technologies to Make Processes and Products More Sustainable Why GAO did this study What GAO found Chemistry contributes to virtually every Stakeholders lack agreement on how to define sustainable chemistry and how to aspect of modern life and the chemical measure or assess the sustainability of chemical processes and products; these industry supports more than 25 percent differences hinder the development and adoption of more sustainable chemistry of the gross domestic product of the technologies. However, based on a review of the literature and stakeholder United States. While these are positive interviews, GAO identified several common themes underlying what sustainable contributions, chemical production can chemistry strives to achieve, including: have negative health and environmental · improve the efficiency with which natural resources—including energy, consequences. -

202 Section: B Course Instructor: Dr BK Singh Study Material (Name of T
Course Name: Methods in Organic Synthesis Paper Number: 202 Section: B Course Instructor: Dr BK Singh Study Material (Name of Topic/Chapter): Organosilicone Compounds, Preparation and applications in organic synthesis; Application of Pd(0) and Pd(II) Complexes in Organic Synthesis- Stille, Suzuki and Sonogashira Coupling, Heck reaction and Negishi Coupling. Dr. BK SINGH, Department of Chemistry, University of Delhi Applications of Pd(0) and Pd(II) complexes in organic synthesis- Heck, Negishi, Suzuki, Stille and Sonogashira Coupling 2010 Nobel Prize in Chemistry awarded jointly to Richard F. Heck, Ei-ichi Negishi, and Akira Suzuki "for palladium-catalyzed cross couplings in organic synthesis" Dr. BK SINGH, Department of Chemistry, University of Delhi Palladium-catalyzed carbon-carbon bond formation via cross coupling The principle of palladium-catalyzed cross couplings is that two molecules are assembled on the metal via the formation of metal-carbon bonds. In this way the carbon atoms bound to palladium are brought very close to one another. In the next step they couple to one another and this leads to the formation of a new carbon-carbon single bond. There are two types of cross-coupling reactions that have become important in organic synthesis. Both reactions are catalyzed by zero valent Pd and both reactions employ an organohalide RX (or analogous compound) as the electrophilic coupling partner. The nucleophilic coupling partner differs in these two reactions. In the first type it is an olefin whereas in the second type it is an organometallic compound R’’M where, M is typically zinc, boron, or tin. Both reactions begin by generating an organopalladium complex RPdX from the reaction of the organic halide with Pd(0). -

Poly(Limonene Carbonate): a Bio-Based & Versatile High-Performance Thermoplastic
Poly(limonene carbonate): a bio-based & versatile high-performance thermoplastic Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. Nat.) an der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth vorgelegt von Oliver Hauenstein aus Dortmund Bayreuth 2016 Die vorliegende Arbeit wurde in der Zeit von November 2012 bis Juni 2016 in Bayreuth am Lehrstuhl Makromolekulare Chemie II unter Betreuung von Herrn Professor Dr. Andreas Greiner angefertigt. Vollständiger Abdruck der von der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth genehmigten Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.). Dissertation eingereicht am: 13.07.2016 Zulassung durch das Leitungsgremium: 26.07.2016 Wissenschaftliches Kolloquium: 01.02.2017 Amtierender Direktor: Prof. Dr. Stephan Kümmel Prüfungsausschuss: Prof. Dr. Andreas Greiner (Erstgutachter) Prof. Dr. Hans-Werner Schmidt (Zweitgutachter) Prof. Dr. Carlo Unverzagt. (Vorsitz) Prof. Dr. Jürgen Senker Drittgutachter: Prof. Dr. Volker Abetz Für meine Familie & Hui Contents Abbreviations & symbols ......................................................................................................................... i List of publications ................................................................................................................................... v Abstract .................................................................................................................................................... -

Recollections of Organopalladium Chemistry*
Pure Appl. Chem., Vol. 71, No. 8, pp. 1539±1547, 1999. Printed in Great Britain. q 1999 IUPAC Recollections of organopalladium chemistry* Jiro Tsuji Emeritus Professor, Tokyo Institute of Technology, Tsu 602±128, Kamakura, Japan Abstract: Organopalladium chemistry has 40 years' history. It was born in late 1950s by the invention of the Wacker process. Inspired by the Wacker reaction, we discovered the ®rst example of the carbon±carbon bond formation by means of a Pd complex, and opened the ®eld of p-allylpalladium chemistry. Historical account of organopalladium chemistry, and major accomplishments in our laboratory in 1960s are presented. INTRODUCTION Invention of the Wacker process to produce acetaldehyde using PdCl2±CuCl2 as catalysts in 1959 is the ®rst example of the homogeneous palladium-catalyzed reaction [1]. Soon after the invention of the Wacker process, formation of vinyl acetate by the oxidative acetoxylation of ethylene with PdCl2 in AcOH in the presence of NaOAc was reported by Moiseev [2]. Vinyl acetate is now produced commercially based on this reaction using supported Pd catalyst in a gas phase (Scheme 1). Scheme 1 As mechanistic explanation of the formation of acetaldehyde, Smidt and co-workers made the following statement in their review [1]. `Nucleophilic attack on ole®n and hydride transfer are characteristic for this reaction. The oxygen required for the oxidation of the ole®n is provided by the water. According to our hypothesis only the oxygen atom of the water is transferred to the ole®n, while the four hydrogen atoms originally present in ethylene remain there.' The early 1960s was the dawn of the research on OMCOS. -

Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View
Review Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View Yulia H. Budnikova *, Yulia B. Dudkina and Mikhail N. Khrizanforov A.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center, Russian Academy of Sciences, Arbuzov str. 8, 420088 Kazan, Russian Federation; [email protected] (Y.B.D.); [email protected] (M.N.K.) * Correspondence: [email protected]; Tel.: +7-843-279-5335 Received: 18 September 2017; Accepted: 12 October 2017; Published: 16 October 2017 Abstract: This review generalizes and specifies the oxidizing ability of a number of oxidants used in palladium (Pd)-catalyzed aromatic C–H functionalizations. The redox potentials have been analyzed as the measure of oxidant strength and applied to the reasoning of the efficiency of known reactions where catalytic cycles include cyclometalated palladium complexes (and other organopalladium key intermediates). Keywords: palladium; oxidant; metal complex catalysis; electrochemistry; redox potential 1. Introduction Transition-metal-catalyzed direct C–H functionalizations open a new road for diverse C–C, C– P, and C–N bond construction in a one-step and atom economical way, without the requirement of prefunctionalized C–H coupling partners [1–6]. The cleavage of high energy C–H bonds (E ~ 110 kcal·mol−1 for C(aryl)–H bonds) typically requires harsh reaction conditions that result in limited substrate scope and low functional group tolerance. The driving force of the current research is the development of the next generation of C–H transformations, which will help make these processes truly useful synthetic tools and develop reactions that proceed under milder conditions. -
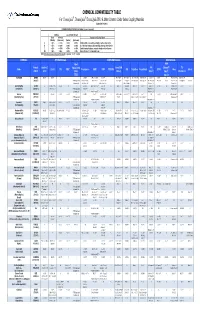
CPC Chemical Compatibility Chart
CHEMICAL COMPATIBILITY TABLE For ChemQuik®, DrumQuik®, DrumQuik PRO & Other Common Colder Series Coupling Materials (Updated 01/14/2010) INTERPRETATION OF TEST DATA (In 30 days to 1 year of exposure) Swelling Loss of Tensile Strength Linear Volumetric Description of Chemical Attack (Plastics) (Elastomers) (Plastics) (Elastomers) A < 10% <= 15% < 15% <=15% Excellent, little or no swelling, softening or surface deterioration B < 15% <= 30% < 30% <= 30% Good chemical resistance, minor swelling, softening or deterioration C < 20% <= 50% < 50% <= 60% Limited chemical resistance, moderate attack, conditional service NR > 20% > 50% > 50% > 60% Severe attack, not recommended for use NOTE: All temperatures are in degrees Fahrenheit. Conversion: °C = (°F - 32)/1.8 CHEMICAL SPRING Materials COUPLING Materials SEAL Materials Teflon® FFKM ® Formula Hastelloy C Encapsulated Acetal/POM FKM (Chemraz / TPO Name 316 SS PPS PEEK™ Polypropylene HDPE PVDF PTFE/PFA ABS Polysulfone Polycarbonate EPDM Buna Silicone (CAS #) (276) 316SS (Celcon) (Viton®) Simriz® / (Santoprene) (TESS) Kalrez®) Acetic Acid C2H4O2 A to 212° 212° A to 212° 212° A A A A to 140° 140° AB to 100% to 70° 70° A to 122° 122° A AAto5%to70 to 5% to 70°° AB 10% to 70° 70° A to 100% to 70° 70° A to 50% to 70° 70° A 10% to 70° 70° AAto70 to 70°° A B to 30% at 70° 70° A to 30% to 70° 70° A (64-19-7) (PTFE Encapsulated AB 50-100% to 160° AB 60% to 180° A to 10% to 225° BC 10% @ 70° C 20% @ 70° A to 20% to 140° B to 50% @ 122° B 10-25% to 100° AB to 200° A to 70° B to 20% to 185° C 50% @ 70° A to -

Chapter 1 the Basic Chemistry of Organopalladium Compounds
Chapter 1 The Basic Chemistry of Organopalladium Compounds 1.1 Characteristic Features of Pd-Promoted or -Catalyzed Reactions There are several features which make reactions involving Pd catalysts and reagents particularly useful and versatile among many transition metals used for organic synthesis. Most importantly, Pd catalysts offer an abundance of possibilities of carbon–carbon bond formation. Importance of the carbon–carbon bond forma- tion in organic synthesis needs no explanation. No other transition metals can offer such versatile methods of the carbon–carbon bond formations as Pd. Tol- erance of Pd catalysts and reagents to many functional groups such as carbonyl and hydroxy groups is the second important feature. Pd-catalyzed reactions can be carried out without protection of these functional groups. Although reactions involving Pd should be carried out carefully, Pd reagents and catalysts are not very sensitive to oxygen and moisture, and even to acid in many reactions catalyzed by Pd–phosphine complexes. It is enough to apply precautions to avoid oxidation of coordinated phosphines, and this can be done easily. On the other hand, the Ni(0) complex is extremely sensitive to oxygen. Of course, Pd is a noble metal and expensive. Its price frequently fluctuates drastically. A few years ago, Pd was more expensive than Pt and Au but cheaper than Rh. As of October 2003, the comparative prices of the noble metals were: Pd (1), Au (1.8), Rh (2.8), Pt (3.3), Ru (0.2). Recently the price of Pd has dropped dramatically, and Pt is currently the most expensive noble metal. Also, the toxicity of Pd has posed no serious problem so far. -

1,1,1,2-Tetrafluoroethane (HFC-134A) (CAS No. 811-97-2) (Second Edition)
1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) JACC No. 50 ISSN-0773-6339-50 Brussels, January 2006 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) ECETOC JACC REPORT No. 50 © Copyright – ECETOC AISBL European Centre for Ecotoxicology and Toxicology of Chemicals 4 Avenue E. Van Nieuwenhuyse (Bte 6), B-1160 Brussels, Belgium. All rights reserved. No part of this publication may be reproduced, copied, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the copyright holder. Applications to reproduce, store, copy or translate should be made to the Secretary General. ECETOC welcomes such applications. Reference to the document, its title and summary may be copied or abstracted in data retrieval systems without subsequent reference. The content of this document has been prepared and reviewed by experts on behalf of ECETOC with all possible care and from the available scientific information. It is provided for information only. ECETOC cannot accept any responsibility or liability and does not provide a warranty for any use or interpretation of the material contained in the publication. ECETOC JACC No. 50 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) CONTENTS EXECUTIVE SUMMARY 1 THE ECETOC SCHEME FOR THE JOINT ASSESSMENT OF COMMODITY CHEMICALS 3 1. SUMMARY AND CONCLUSIONS 4 2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS 6 2.1 Identity 6 2.2 EU classification and labelling 6 2.3 Physical and chemical properties 6 2.4 Conversion factors 8 2.5 Analytical methods 8 3.