Management of Patients with Viral Hepatitis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Forgotten F1 Teams – Series 1 Omnibus Simtek Grand Prix
Forgotten F1 Teams – Series 1 Omnibus Welcome to Forgotten F1 Teams – a mini series from Sidepodcast. These shows were originally released over seven consecutive days But are now gathered together in this omniBus edition. Simtek Grand Prix You’re listening to Sidepodcast, and this is the latest mini‐series: Forgotten F1 Teams. I think it’s proBaBly self explanatory But this is a series dedicated to profiling some of the forgotten teams. Forget aBout your Ferrari’s and your McLaren’s, what aBout those who didn’t make such an impact on the sport, But still have a story to tell? Those are the ones you’ll hear today. Thanks should go to Scott Woodwiss for suggesting the topic, and the teams, and we’ll dive right in with Simtek Grand Prix. Simtek Grand Prix was Born from Simtek Research Ltd, the name standing for Simulation Technology. The company founders were Nick Wirth and Max Mosley, Both of whom had serious pedigree within motorsport. Mosley had Been a team owner Before with March, and Wirth was a mechanical engineering student who was snapped up By March as an aerodynamicist, working underneath Adrian Newey. When March was sold to Leyton House, Mosley and Wirth? Both decided to leave, and joined forces to create Simtek. Originally, the company had a single office in Wirth’s house, But it was soon oBvious they needed a Bigger, more wind‐tunnel shaped Base, which they Built in Oxfordshire. Mosley had the connections that meant racing teams from all over the gloBe were interested in using their research technologies, But while keeping the clients satisfied, Simtek Began designing an F1 car for BMW in secret. -

FINAL -SIRS-Program-Book-2018-Online.Pdf
6th Schizophrenia International Research Society Conference Integrated Prevention and Treatment: Shifting the Way We Think FLORENCE, ITALY 4 – 8 APRIL 2018 6th Schizophrenia International Research Society Conference Integrated Prevention and Treatment: Shifting the Way We Think Opening Letter Dear Attendees, It is our great pleasure to welcome you to the 6th Biennial Schizophrenia International Research Society (SIRS) Conference. SIRS is a non-profit organization dedicated to promoting research and communication about schizophrenia among research scientists, clinicians, drug developers, and policy makers internationally. We sincerely appreciate your interest in the Society and in our conference. The fifth congress in 2016 was a major success for the field attracting more than 1800 attendees from 52 countries. We anticipate an even higher attendance at this congress with most of the best investigators in the world in attendance. SIRS was founded in 2005 with the goal of bringing together scientists from around the world to exchange the latest advances in biological and psychosocial research in schizophrenia. The Society is dedicated to facilitating international collaboration to discover the causes of, and better treatments for, schizophrenia and related disorders. Part of the mission of the Society is to promote educational programs in order to effectively disseminate new research findings and to expedite the publication of new research on schizophrenia. The focus of the 6th Biennial Conference is ‘Integrated Prevention and Treatment: Shifting the Way We Think’. Under the outstanding leadership of Program Committee Chairs, Paola Dazzan, Bita Moghaddam, and Eóin Killackey, we have an exciting scientific program planned for this year. The Program Committee selected thirty-five outstanding symposia sessions out of 103 submissions. -
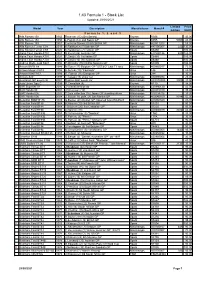
Stock List Updated 28/09/2021
1:43 Formula 1 - Stock List Updated 28/09/2021 Limited Price Model Year Description Manufacturer Manuf # Edition (AUD) F o r m u l a 1 , 2 a n d 3 Alfa Romeo 158 1950 Race car (25) (Oro Series) Brumm R036 35.00 Alfa Romeo 158 1950 L.Fagioli (12) 2nd Swiss GP Brumm S055 5000 40.00 Alfa Romeo 159 1951 Consalvo Sanesi (3) 6th British GP Minichamps 400511203 55.00 Alfa Romeo Ferrari C38 2019 K.Raikkonen (7) Bahrain GP Minichamps 447190007 222 135.00 Alfa Romeo Ferrari C39 2020 K.Raikkonen (7) Turkish GP Spark S6492 100.00 Alpha Tauri Honda AT01 2020 D.Kvyat (26) Austrian GP Minichamps 417200126 400 125.00 Alpha Tauri Honda AT01 2020 P.Gasly (10) 1st Italian GP Spark S6480 105.00 Alpha Tauri Honda AT01 2020 P.Gasly (10) 7th Austrian GP Spark S6468 100.00 Andrea Moda Judd S921 1992 P.McCathy (35) DNPQ Monaco GP Spark S3899 100.00 Arrows BMW A8 1986 M.Surer (17) Belgium GP "USF&G" Last F1 race Minichamps 400860017 75.00 Arrows Mugen FA13 1992 A.Suzuki (10) "Footwork" Onyx 146 25.00 Arrows Hart FA17 1996 R. Rosset (16) European GP Onyx 284 30.00 Arrows A20 1999 T.Takagi (15) show car Minichamps 430990084 25.00 Australian GP Event car 2001 Qantas AGP Event car Minichamps AC4010300 3000 40.00 Auto Union Tipo C 1936 R.Gemellate (6) Brumm R110 38.00 BAR Supertec 01 2000 J.Villeneuve test car Minichamps 430990120 40.00 BAR Honda 03 2001 J.Villeneuve (10) Minichamps 400010010 35.00 BAR Honda 005 2003 T.Sato collection (16) Japan GP standing driver Minichamps 518034316 35.00 BAR Honda 006 2004 J. -

January 2013 Available FORMULA 1 Cars
January 2013 available FORMULA 1 cars Arrows A11B Cosworth/ Hart 650 bhp DFR engine year 1990 The car has just had a rebuild with a newly installed ATL new fuel cell and oil cooler and freshly rebuilt Hart version of the DFR 650 bhp engine with £20k invoice This car is ideal for Euroboss events or F1 track day demo events Car has not been used since re assembly Arrows FA16 1994 car with empty Hart 3 litre V8 engine This car was raced to 3rd place at Australian GP in 94 by Gianni Morbidelli The car has sat since its last GP in the private collection of Arrows F1 team owner until it was purchased by the current owner The car can be rebuilt to race in Eurpboss events with the Hart V8 motor ( parts are obtainable) or fitted with Judd V10 or Cosworth HB V8 engines as fitting kits are available The gearbox is semi automatic unit but can be run as a manual sequential garbo BAR F1 show car built with glass fibre chassis and many real parts ie rear wing bodywork some suspension parts Ideal for static display/promotional work Currently painted in a paint scheme for present promotional work Copersucar F6A Emerson Fittipaldi Rolling chassis Car is 100% complete less engine but a choice of fresh rebuilt long and short stroke DFV engines are available This is the last F1 car that Emerson raced before the retired from F1 This car has not raced since 1994 having sat in private collection Suitable to race in Historic F1 in UK/EEC USA series will consider interesting trades Footwork Arrows A14 this car raced in the 1993 F1 World Championship with Mugen V10 engines but they are not available and this example has been fitted with Hart DFR 3.5 litre engine. -

Guide Média 2017 2017 Media Kit Table Des Matières | Table of Contents
GUIDE MÉDIA 2017 2017 MEDIA KIT TABLE DES MATIÈRES | TABLE OF CONTENTS SECTION 1 : RENSEIGNEMENTS GÉNÉRAUX | GENERAL INFORMATION COMITÉ ORGANISATEUR | ORGANIZING COMMITTEE 4 À PROPOS | A FEW FACTS 5 SECTION : 2 SERVICES DE PRESSE | MEDIA SERVICES PERSONNEL DU CENTRE MÉDIA | MEDIA CENTRE STAFF 7 CENTRE MÉDIA | MEDIA CENTRE 8 CENTRE D’ACCRÉDITATION MÉDIA | MEDIA ACCREDITATION CENTRE 9 NAVETTES MÉDIA | MEDIA SHUTTLES 9 CONFÉRENCES DE PRESSE | PRESS CONFERENCES 10 SERVICES AUX PHOTOGRAPHES | SERVICES FOR PHOTOGRAPHERS 11 HALTE MÉDIA | MEDIA HOSPITALITY 12 CARTE DU SITE | SITE MAP 13 CARTE DU CENTRE MÉDIA | MEDIA CENTRE MAP 14 ALLOCATION DES STANDS ET DES AIRES D’HOSPITALITÉ | PITS AND HOSPITALITIES ALLOCATION 14 SECTION : 3 CHAMPIONNAT DU MONDE DE FORMULE 1 FIA 2017 | 2017 FIA FORMULA 1 WORLD CHAMPIONSHIP LISTE DES ENGAGÉS 2017 | 2017 ENTRY LIST 17 CLASSEMENT PROVISOIRE DES PILOTES ET CONSTRUCTEURS | PROVISIONAL DRIVER & CONSTRUCTOR STANDINGS 18 POLES, VICTOIRES ET MEILLEURS TOURS | POLES, WINNERS & BEST LAPS 18 POINTAGE DES PILOTES LORS DES 6 PREMIÈRES RONDES | DRIVER STANDINGS AFTER 6 ROUNDS 19 POINTAGE DES CONSTRUCTEURS LORS DES 6 PREMIÈRES RONDES | CONSTRUCTOR STANDINGS AFTER 6ROUNDS 20 RÉSULTATS DES COURSES | RACE RESULT 21 SECTION : 4 ÉQUIPES DE FORMULE 1 | FORMULA 1 TEAMS 27 SECTION : 5 FORMULA 1 GRAND PRIX DU CANADA STATISTIQUES | STATISTICS 46 LES 6-8-10 PREMIERS DEPUIS 1967 | TOP 6-8-10 SINCE 1967 54 SECTION : 6 PERFORMANCES DES DERNIÈRES ANNÉES | RECENT YEARS’ RECORDS CLASSEMENT DES PILOTES 2014-2016 | 2014-2016 DRIVERS’ STANDING 59 SECTION : 7 COURSES DE SOUTIEN | SUPPORT RACES CHALLENGE FERRARI TROFEO PIRELLI | TROFEOPIRELLIFERRARICHALLENGE 61 ULTRA 94 PORSCHE GT3 CUP CHALLENGE CANADA BY YOKOHAMA. -

Download the Official 2002/2003 Formula 1 Annual
The Official 2002/2003 Formula 1 Annual, Bernie Ecclestone, Formula One Publishing, Formula One Publishing, 2003, 0954414705, 9780954414702, . DOWNLOAD HERE Life of Senna The Biography of Ayrton Senna, Tom Rubython, May 1, 2004, , 602 pages. Inside the Mind of the Grand Prix Driver The Psychology of the Fastest Men on Earth: Sex, Danger and Everything Else, Christopher Hilton, Jan 18, 2004, , 384 pages. This book gets behind the PR-speak to explore what Grand Prix drivers really think and feel. They talk with rare frankness of their hopes and fears, how they began racing and .... The Piranha Club Power and Influence in Formula One, Timothy Collings, Sep 1, 2004, Sports & Recreation, 336 pages. Now fully updated, "The Piranha Club" is the first serious study of Formula One's most intriguing and influential figures - the men who wield the real power. It is an .... Formula One Yearbook 2003-2004 , Luc Domenjoz, Jan 1, 2004, , 224 pages. The International Grand Prix annual, and the essential reference guide to the 2003 Grand Prix season. Many articles, anecdotes and analysis. Complete and detailed results of .... Flat Out & Flat Broke Formula 1 the Hard Way, Perry L. McCarthy, Sep 1, 2002, , 256 pages. Perry McCarthy decided at the age of 18 that he wanted to reach the top in the world's most expensive sport. This is his autobiography.. Juan Pablo Montoya , Christopher Hilton, May 1, 2003, , 144 pages. 'There's an expression in the United States 'someone's got iced water in their veins' and that's how he drives: take no prisoners, don't give a damn, don't be intimidated by ... -

Manual, Accompanying Documentation and CD Are Copyrighted
An Official Product of the FIA Formula One World Championship Licensed by FOCA to Fuji Television Game Copyright © 1996 Edward Grabowski Communications Ltd Packaging, Documentation and Logo © 1996 MicroProse This manual, accompanying documentation and CD are copyrighted. The owner of this product is entitled to use this product for his or her own use. Except for the quoting of brief passages for the purposes of reviews, no one may transfer, copy, back-up, give or sell any part of the manual or the information on the CD, or transmit in any form or by any means, electronic, mechanical, photocopying, recording or otherwise without the prior permission of the publisher. Any person or persons reproducing any part of this program, in any media, for any reason, shall be guilty of copyright violation and shall be subject to civil liability at the discretion of the copyright holder. Not for rent or hire. Made in the UK. 2 CAREER OPTIONS . .28 INTRODUCTION . .5 Quickstart . .28 A BRIEF WORD FOR Long Term Contract . .28 GRAND PRIX MANAGER VETERANS . .5 Short Term Contract . .28 FINDING OUT WHERE EVERYTHING IS . .5 Link Play . .29 MAIN MENU SCREEN . .29 GETTING STARTED . .6 PERSONNEL . .29 Contents INSTALLATION AND LOADING . .6 Chief Designer . .29 THE POINT-AND-CLICK INTERFACE . .7 Chief Engineer . .30 Mouse Control . .7 Chief Mechanic . .32 Keyboard Control . .7 Commercial Manager . .32 THE MANUAL . .7 Drivers . .32 QUICK START TUTORIAL . .8 Injured Drivers . .33 MANAGEMENT TUTORIAL . .10 R&D . .33 Organising Grand Prix Manager 2 . .10 New Car Chassis . .33 Planning your Triumph . .10 Acquire Technology . -

Forgotten F1 Teams – Arrows
Forgotten F1 Teams – Arrows Welcome to Forgotten F1 Teams – a mini series from Sidepodcast chronicling the rise and fall of teams that couldn’t stay the distance. This is the fourth show, we’ve already looked at Simtek, Pacific and Forti, but today we’re talking about Arrows. Arrows Grand Prix came to life in 1977, founded by previous Shadow employees, Franco Ambrosio, Alan Rees, Jackie Oliver, Dave Wass and Tony Southgate – the surnames of whom spell out Arrows… sort of. Based in Milton Keynes, their Formula 1 entry was a bit of a rush job, with their first ever car being produced in just 53 days. Riccardo Patrese was brought on board to steer the car and the early signs were promising. They finished tenth in their first race in ‘78, almost won their second, and picked up some points in their third Grand Prix. Unfortunately, the team didn’t have an easy start to life with some early controversies. Founder Franco Ambrosio had to leave the team after being found guilty of financial misconduct in Italy, and sent to prison. The team were then sued by Shadow for copyright on their chassis, which was upheld by the court. However, this didn’t stop them, as while the decision was being made over the the copied chassis, Arrows had built a brand new car, which then took to the track without them having to miss a race. Patrese was involved in a serious multi‐car pile up at the Italian Grand Prix that year. Fellow driver Ronnie Peterson died after the accident, and James Hunt, who was also involved, led a successful campaign to get Patrese banned from racing at the next Grand Prix. -

LISTE DES ENGAGES Première Page À Demander Auprès De Héléna Ou Jérémy De L'aco
LE MANS 2018 - 86 ème édition InfosCourse LISTE DES ENGAGES première page à demander auprès de Héléna ou Jérémy de l'ACO 1 REBELLION RACING InfosCourse Au Mans : 9 Part : 3 Vict + 2 Pod ; 1 Pole & 8°Pro-Cup 24h Spa /AudiR8 TOM’s,1 Vict + 4 Pod. LOTTERER André LMS WRT 08 à 09 A1 GP /Team D,, 4 MT F1 / Caterham Renault, 08 3°F.Nippon / TOMs, 4 pod. 17 AB /P.919 Hybrid Porsche Audi Joest (Fässler, Tre- FFSA GT /Audi R8BAC Team 3°SuperGT 500 /Lexus (Jani, Tandy) luyer) WRT , Vict Spa TOMs, 4 pod. 16 4° /Audi R18 Audi Joest 12 Vict & PP /Audi R18 e-tron 13 Vice-Chpion WECP1 /Audi 07 5°F.Nippon / TOMs , 1 vict + (Fässler, Treluyer) Audi Joest (Treluyer, Fäss- R18 A.S.Team Joest, 3 Vict 2 pod. 15 3° & MT /Audi R18 e-tron ler) 2° Silverstone & Bahrain, 6°Super GT500/ Lexus Toms, Audi Joest (Fässler, Tre- 11 Vict & MT /Audi R18 Audi 3°Austin 4 pod. luyer) Joest (Treluyer, Fässler) Vice-Chpion Japan SF / Team 06 Chpion Super GT500 /Lexus- 14 Vict & MT /Audi R18 e-tron 10 2° /Audi R15 Audi Joest Tom’s, 2 Vict Tom’s, 1 Vict +2 Pod. DEU Audi Joest (Fässler, Tre- (Fässler, Treluyer) 3°24h Spa/ Audi R8 3°F. Nippon /Tom’s , 2 vict luyer) 09 7° /Audi R10 Kolles (Charles 12 Chpion WEC P1 /Audi R18 e- 05 4°F.Nippon/Nakajima 2 vict 13 5° & MT /Audi R18 e-tron Jr Zwolsman, Karthikeyan) tron A. S. Team Joest, 3 Vict 17°Super GT500 / NSX 4° F. -

BOSS GP Peroni Race Weekend - 2 & 3 July - Autodromo Nazionale Monza
BOSS GP Peroni Race Weekend - 2 & 3 July - Autodromo Nazionale Monza No. Team Driver Nat. Make Type Engine Class 1 Team Ascari Klaas Zwart NED Jaguar R5 F1 Cosworth 3.0 V10 OPEN 2 Top Speed Ingo Gerstl AUT Toro Rosso STR1 F1 Cosworth 3.0 V10 OPEN 3 GP Racing Peter Milavec AUT Panoz DP01 Champcar Judd 4.2 V10 OPEN 5 H&A Racing Wolfgang Jordan GER Benetton B197 F1 Judd 4.0 V10 OPEN 10 Ves Racing Frits van Eerd NED Minardi PS04B F1 Cosworth 3.0 V10 OPEN 21 H&A Racing Bernd Herndlhofer AUT Benetton B197 F1 Judd 4.0 V10 OPEN 22 Speed Center - Castrol Hans Laub GER Forti FG03 F1 Judd 4.0 V10 OPEN 25 H&A VOX Racing Lance David Arnold GER Benetton B197 F1 Judd 4.0 V10 OPEN 100 Top Speed Chris Höher AUT Dallara GP2 Mecachrome 4.0 V8 FORMULA 101 Speed Center - Castrol Peter Göllner SUI Dallara GP2 Mecachrome 4.0 V8 FORMULA 102 Racing Experience Stuart Wiltshire GBR Dallara GP2 Mecachrome 4.0 V8 FORMULA 103 Racing Experience - Penn Elcom Phil Stratford USA Dallara GP2 Mecachrome 4.0 V8 FORMULA 110 Hoffmann Racing Walter Steding GER Dallara GP2 Mecachrome 4.0 V8 FORMULA 115 PS Racing Mahaveer Raghunathan IND Lola B05/52 Auto GP Gibson Tech 3.4 V8 FORMULA 117 Easy Formula Christopher Brenier FRA Panoz DP09B Superleague Formula Menard CT 4.2 V12 FORMULA 118 Easy Formula Gilles Brenier FRA Panoz DP09B Superleague Formula Menard CT 4.2 V12 FORMULA 122 Inter Europol Competition Jens Renstrup DEN Dallara GP2 Mecachrome 4.0 V8 FORMULA 126 Griffith's Bruno Navarrete FRA Dallara GP2 Mecachrome 4.0 V8 FORMULA 136 De Boer Manx Henk de Boer NED Dallara GP2 Mecachrome -

Alternative Formats If You Require This Document in an Alternative Format, Please Contact: [email protected]
University of Bath PHD Tacit Knowledge Transfer in Inter-Organisational Networks A social network analysis of Formula 1 Mishra, Danish Award date: 2018 Awarding institution: University of Bath Link to publication Alternative formats If you require this document in an alternative format, please contact: [email protected] General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 06. Oct. 2021 Tacit Knowledge Transfer in Inter-Organisational Networks: A social network analysis of Formula 1 Danish Mishra A thesis submitted for the degree of Doctor of Philosophy Department of Mechanical Engineering University of Bath September 2017 Copyright Attention is drawn to the fact that copyright of this thesis rests with its author. A copy of this thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and they must not copy it or use material from it except as permitted by law or with the consent of the author. -

The 1997 Concorde Agreement Bringing Transparency to Formula 1
The 1997 Concorde Agreement Bringing Transparency to Formula 1 It is Formula 1’s holy of holies, the secret kept by scores contracts written by lawyers can be. Much of it — like of people for a quarter of a century. It is the contract the driver recognition board agreement, which occupies which binds the F1 teams, the FIA, and the entity, run by 25 of 103 pages — has little utility for those seeking a Bernie Ecclestone, which holds the sport’s commercial deeper understanding of the machinations of Formula 1. rights through 2097. It is the fabled Concorde A much smaller portion — the sections on the Formula Agreement, and now, it is secret no longer. One Commission and the Formula One Technical In truth, we never expected to see the Concorde, even Working Group, for example — is essential to after, as seems likely now, it might pass into history at understanding such things as how Mosley brought forth the end of 2007, to be replaced by a very different the 2005 regulation changes under the guise of safety. contract. But then, on an otherwise unremarkable day These and other sections are also essential to nearly eight years ago, a heavy, nondescript package understanding the dispute between the FIA and the arrived in the post. No return address. No hint at the Grand Prix Manufacturers Assn. contents. To our considerable surprise, opening the A very small portion is fascinating. But on balance, packaged produced three thick, bound documents: the we suspect that, when the reader has finished plowing 1992 Concorde Agreement, the 1994 Variation through it, the reaction will be the same as ours: why Agreement to the 1992 Concorde Agreement, and the did the players feel it necessary to keep this secret? 1997 Concorde Agreement, then the most recent signed Perhaps it was simply to make them feel important, and by the teams, the FIA and Ecclestone.