Development of Novel Methods and Applications in Total Synthesis of Natural Products
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And C,N-Chelated Organocopper Compounds†
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 September 2021 Review C,C- and C,N-Chelated Organocopper Compounds† Liang Liu1, Hui Chen2, Zhenqiang Yang2, Junnian Wei1* and Zhenfeng Xi1,3* 1 Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China; [email protected] (L.L.), [email protected] (J.W.), [email protected] (Z.X.) 2 Henan Institute of Chemistry Co. Ltd., Henan Academy of Sciences, Zhengzhou 450002, China; [email protected] (H.C.), [email protected] (Z.Y.) 3 State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China; [email protected] (Z.X.) * Correspondence: [email protected] (J.W.), [email protected] (Z.X.) † Dedicated to Professor Gerard van Koten on the occasion of his 80th birthday Abstract: Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure- function relationship of organocopper compounds would advance rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono- carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize the unstable organocopper compounds. The bidentate ligands can chelate to the same copper atom via 휂2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via 휂1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. -

Conversion of Carbon Dioxide to Acetylene on a Micro Scale
810 NATURE June 14, 1947 Vol. 159 orbitale of the ethmoid is reduced". In the orang Stainless steel was found to be the most satis and the gibbon a large planum orbitale articulates factory furnace material tried. From mild steel in front with the lacrimal, as in man. The figure relatively large amounts of acetylene were produced we give of the orbital wall in Pleaianthropus shows in blank experiments, and a fused silica envelope a condition almost exactly as in man. fitted with a nickel thimble was found, after it had We are here not at present concerned with the been used with calcium and barium metals, to absorb question of whether man and the Australopithecinre carbon dioxide when hot even when no calcium or have arisen from an early anthropoid, or a pre barium was present. In carrying out the absorption anthropoid, or an Old World monkey or a tarsioid ; of carbon dioxide by barium metal in the stainless but we think the evidence afforded by this new skull steel furnace it was found that when the pressure of Plesianthropus shows that the Australopithecinre at which the gas was admitted was less than about and man are very closely allied, and that these small 10·1 mm. of mercury, the yield of acetylene was brained man-like beings were very nearly human. variable and only about 45 per cent. Good yields R. BROOM were obtained when the carbon dioxide at its full J. T. RoBINSON pressure was admitted to the furnace before raising Transvaal Museum, Pretoria. the temperature above 400° C. -
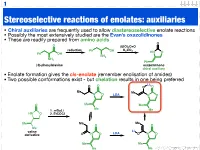
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Nomenclature of Carboxylic Acid Derivatives Acid Halide Substituents
Gentilucci, Carboxylic Acid Derivatives Nomenclature of Carboxylic Acid Derivatives Gentilucci, Carboxylic Acid Derivatives Acid halides 1. Alkane + the suffix -oyl followed by the halogen. 2. Select the longest continuous carbon chain, containing the acyl group. 3. Number the carbon chain, beginning at the end nearest to the acyl group. 4. Number the substituents and write the name, listing substituents alphabetically. Acid halide substituents attached to rings are named using the suffix - carbonyl. 1 Gentilucci, Carboxylic Acid Derivatives Anhydrides 1. Symmetrical: replace the ending "acid" with "anhydride ". 2. Asymmetrical: select the longest continuous carbon chain, containing the carboxylic acid group, and derive the parent name by replacing the -e ending with -oic anhydride . 3. Number the carbon chain, beginning at the end nearest to the acyl group. 4. Number the substituents and write the name, listing substituents alphabetically. Gentilucci, Carboxylic Acid Derivatives Amides are named by replacing the ending -oic acid with -amide . 1. Select the longest continuous carbon chain, containing the acyl group, and derive the parent name by replacing the -e ending with -amide . 2. Number the carbon chain, beginning at the end nearest to the acyl group. 3. Number the substituents and write the name, listing substituents alphabetically. 4. If the nitrogen atom is further substituted, the substituents are preceded by N- to indicate that they are attached to the nitrogen. Acid halide substituents attached to rings are named using the suffix - carboxamide. 2 Gentilucci, Carboxylic Acid Derivatives Carboxylate esters 1. Select the longest continuous carbon chain containing the acyl group, and derive the parent name by replacing the -e ending with –oate . -

Carbonyl Compounds
Carbonyl Compounds What are Carbonyl Compounds? Carbonyl compounds are compounds that contain the C=O (carbonyl) group. Preparation of Aldehydes: 1. Preparation from Acid Chloride (Rosenmund Reduction): This reaction was named after Karl Wilhelm Rosenmund, who first reported it in 1918. The reaction is a hydrogenation process in which an acyl chloride is selectively reduced to an aldehyde. The reaction, a hydrogenolysis, is catalysed by palladium on barium sulfate, which is sometimes called the Rosenmund catalyst. 2. Preparation from Nitriles: This reaction involves the preparation of aldehydes (R-CHO) from nitriles (R- CN) using SnCl2 and HCl and quenching the resulting iminium salt ([R- + − CH=NH2] Cl ) with water (H2O). During the synthesis, ammonium chloride is also produced. The reaction is known as Stephen Aldehyde synthesis. Dr. Sumi Ganguly Page 1 3. Preparation from Grignard Reagent: When Grignard Reagent is reacted with HCN followed by hydrolysis aldehyde is produced. Preparation of Ketones: 1. Preparation from Acid Chloride (Friedel-Crafts Acylation): Acid chlorides when reacted with benzene in presence of anhydrous AlCl3, aromatic ketone are produced. However, only aromatic ketones can be prepared by following this method. In order to prepare both aromatic and aliphatic ketones acid chlorides is reacted with lithium dialkylcuprate (Gilman Reagnt). Dr. Sumi Ganguly Page 2 The lithium dialkyl cuprate is produced by the reaction of two equivalents of the organolithium reagent with copper (I) iodide. Example: 3. Preparation from Nitriles and Grignard Reagents: When Grignard Reagent is reacted with RCN followed by hydrolysis aldehyde is produced. Dr. Sumi Ganguly Page 3 Physical Characteristic of Carbonyl Compounds: 1) The boiling point of carbonyl compounds is higher than the alkanes with similar Mr. -

Acetal (POM) Chemical Compatibility Chart From
ver 31-Mar-2020 Acetal (POM) Chemical Compatibility Chart Chemical Chemical Acetaldehyde A Ammonium Acetate C Acetamide A Ammonium Bifluoride D Acetate Solvents A Ammonium Carbonate D Acetic Acid D Ammonium Caseinate D Acetic Acid, 20% C Ammonium Chloride, 10% B Acetic Acid, 80% D Ammonium Hydroxide D Acetic Acid, Glacial D Ammonium Nitrate, 10% A Acetic Anhydride D Ammonium Oxalate B Acetone A Ammonium Persulfate D Acetyl Chloride, dry D Ammonium Phosphate, Dibasic B Acetylene A Ammonium Phosphate, Monobasic B Alcohols: Amyl A Ammonium Phosphate, Tribasic B Alcohols: Benzyl A Ammonium Sulfate B Alcohols: Butyl A Ammonium Sulfite D Alcohols: Diacetone A Ammonium Thiosulfate B Alcohols: Ethyl A Amyl Acetate B Alcohols: Hexyl A Amyl Alcohol A Alcohols: Isobutyl A Amyl Chloride A Alcohols: Isopropyl A Aniline A Alcohols: Methyl A Aniline Oil D Alcohols: Octyl A Anise Oil D Alcohols: Propyl (1-Propanol) A Antifreeze D Aluminum chloride, 20% C Aqua Regia (80% HCl, 20% HNO3) D Aluminum Fluoride C Aromatic Hydrocarbons A Aluminum Hydroxide A Arsenic Acid D Aluminum Nitrate B Asphalt B Aluminum Potassium Sulfate, 10% C Barium Carbonate A Aluminum Potassium Sulfate, 100% C Barium Chloride A Aluminum Sulfate, 10% B Barium Cyanide B Alums C Barium Hydroxide D Amines D Barium Nitrate B Ammonia, 10% (Ammonium Hydroxide) C Barium Sulfate B Ammonia, 10% D Barium Sulfide A Ammonia, anhydrous D Bay Oil D Ammonia, liquid D Beer A Ammonia Nitrate C Beet Sugar Liquids B Key to General Chemical Resistance – All data is based on ambient or room temperature conditions, about 64°F (18°C) to 73°F (23°C) A = Excellent C = Fair - Moderate Effect, not recommended B= Good - Minor Effect, slight corrosion or discoloration D = Severe Effect, not recommended for ANY use It is the sole responsibility of the system designer and user to select products suitable for their specific application requirements and to ensure proper installation, operation, and maintenance of these products. -
![INDOLES from 2-METHYLNITROBENZENES by CONDENSATION with FORMAMIDE ACETALS FOLLOWED by REDUCTION: 4-BENZYLOXYINDOLE [1H-Indole, 4-(Phenylmethoxy)-]](https://docslib.b-cdn.net/cover/2671/indoles-from-2-methylnitrobenzenes-by-condensation-with-formamide-acetals-followed-by-reduction-4-benzyloxyindole-1h-indole-4-phenylmethoxy-222671.webp)
INDOLES from 2-METHYLNITROBENZENES by CONDENSATION with FORMAMIDE ACETALS FOLLOWED by REDUCTION: 4-BENZYLOXYINDOLE [1H-Indole, 4-(Phenylmethoxy)-]
DOI:10.15227/orgsyn.063.0214 Organic Syntheses, Coll. Vol. 7, p.34 (1990); Vol. 63, p.214 (1985). INDOLES FROM 2-METHYLNITROBENZENES BY CONDENSATION WITH FORMAMIDE ACETALS FOLLOWED BY REDUCTION: 4-BENZYLOXYINDOLE [1H-Indole, 4-(phenylmethoxy)-] Submitted by Andrew D. Batcho1 and Willy Leimgruber2. Checked by David J. Wustrow and Andrew S. Kende. 1. Procedure A. 6-Benzyloxy-2-nitrotoluene. A stirred mixture of 124.7 g (0.81 mol) of 2-methyl-3-nitrophenol (Note 1), 113.2 g (0.90 mol) of benzyl chloride, 112.2 g (0.81 mol) of anhydrous potassium carbonate, and 800 mL of dimethylformamide (DMF) is heated at 90°C for 3 hr. Most of the DMF is removed on a rotary evaporator (20 mm) and the oily residue is poured into 400 mL of 1 N sodium hydroxide and extracted with ether (3 × 800 mL). The combined extracts are dried (Na2SO4), filtered, and evaporated to give 203.5 g of yellowish solid. Recrystallization from 1 L of methanol cooled to 0°C affords 177.6 (90%) of 6-benzyloxy-2-nitrotoluene as pale-yellow crystals, mp 61–63°C3 (Note 2). B. (E)-6-Benzyloxy-2-nitro-β-pyrrolidinostyrene. To a solution of 175.4 g (0.72 mol) of 6- benzyloxy-2-nitrotoluene in 400 mL of DMF are added 102.5 g (0.84 mol) of N,N-dimethylformamide dimethyl acetal (Note 3) and 59.8 g (0.84 mol) of pyrrolidine. The solution is heated at reflux (110°C) for 3 hr (Note 4) under nitrogen and allowed to cool to room temperature. -

European Journal of Biomedical and Pharmaceutical Sciences
ejbps, 2016, Volume 3, Issue 1, 62-83. Review Article SJIF Impact Factor 2.062 Kumar et al. European Journal European ofJournal Biomedical of Biomedical and Pharmaceutical ISSNSciences 2349 -8870 Volume: 3 AND Pharmaceutical sciences Issue: 1 62-83 http://www.ejbps.com Year: 2016 A REVIEW ON THE PHYTOCONSTITUENTS AND PHARMACOLOGICAL ACTIONS IN THE MEDICINAL PLANTS OF BEDABUNA FOREST, JIMMA ZONE, SOUTH WEST ETHIOPIA REPORTED EFFECT ON EXPERIMENTAL MODELS Kumar Ganesan1*, Suresh Kumar P. Nair1, Melese Sinaga1, Sharmila Banu Gani2* 1Department of Biomedical Sciences, School of Public Health and Medical Sciences, Jimma University, Jimma 378, Ethiopia 2Department of Zoology, NKR Government Arts College for Women, Namakkal-637001, Tamilnadu, India *Author for Correspondence: Dr. Kumar Ganesan Department of Biomedical Sciences, School of Public Health and Medical Sciences, Jimma University, Jimma 378, Ethiopia Article Received on 03/11/2015 Article Revised on 24/11/2015 Article Accepted on 15/12/2015 ABSTRACT Ethiopia is sixth largest biodiversity centre in the world having numerous ethinic cultures, climate and topographies. The present paper reviews on medicinal properties along with atypical Phytoconstituents and pharmacological actions of various plants in bedabuna forest, Zimma zone, Southwest Ethiopia, which has been reported effect on experimental models. This study is very authentic and helpful to find richest bioresources like identification of medicinal plants, documentation, protection and sustainable usages. This study will helpful to not only a native people of Jimma, southwest Ethiopia but also the other part of the Ethiopia to explore the indigenous medicinal plants used in the treatment of various ailments for human and livestock. In the present study totally 49 species of traditional medicinal plants belonging to 31 families were come across by regular ground visits and arbitrarily interviewed with native participants. -

The Total Synthesis of Securinine and Other Methodology Studies
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 2010 The total synthesis of securinine and other methodology studies Bhartesh Dhudshia University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Dhudshia, Bhartesh, "The total synthesis of securinine and other methodology studies" (2010). Electronic Theses and Dissertations. 8275. https://scholar.uwindsor.ca/etd/8275 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. The Total Synthesis of Securinine and Other Methodology Studies by Bhartesh Dhudshia A Dissertation Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry in Partial Fulfillment of the Requirements -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Poly( Ethylene-Co-Methyl Acrylate)-Solvent- Cosolvent Phase Behaviour at High Pressures
Poly( ethylene-co-methyl acrylate)-solvent- cosolvent phase behaviour at high pressures Melchior A. Meilchen, Bruce M. Hasch, Sang-Ho Lee and Mark A. McHugh* Department of Chemical Engineering, Johns Hopkins University, Baltimore, MD 21278, USA (Received 5 April 7991; accepted 75 July 1997 ) Cloud-point data to 160°C and 2000 bar are presented showing the effect of cosolvents on the phase behaviour of poly(ethylene-co-methyl acrylate) (EMA) (64 mo1%/36 mol%) with propane and chlorodifluoromethane (F22). Ethanol shifts the EMA-propane cloud-point curves to lower temperatures and pressures, but above _ 10 wt% ethanol, the copolymer becomes insoluble. Up to 40 wt% acetone monotonically shifts the EMA-propane cloud-point curves to lower temperatures and pressures. Acetone and ethanol both shift the cloud-point curves of EMA-F22 mixtures in the same monotonic manner for cosolvent concentrations of up to 40 wt%. (Keywords: copolymer; cosolvent; high pressures) INTRODUCTION or diethyl ether, so it is necessary to operate at elevated temperatures and pressures to dissolve high molecular Within the past 20 years there has been a great deal of weight polystyrene in either solvent. Adding acetone to effort invested in trying to understand and model the PS-diethyl ether mixtures monotonically reduces the solubility behaviour of polar polymers in liquid cosolvent pressure needed at a given temperature to obtain a single mixtures’-9. Polymer solubility is usually related to phase. whether the cosolvent preferentially solvates or adsorbs There has also been a number of viscometric and light to certain segments of the polymer chain as determined scattering studies on the solution behaviour of polar by light scattering, viscosity measurements or cloud- copolymers in liquid cosolvent mixtures’4*‘5.