Organic Chemistry 12Th Wiley
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemicals, Rare Chemicals, Reagents & Solvents
Annual Procurement Plan for the Year 2021-22 Consumable List 1 Chemicals, Rare Chemicals, Reagents & Solvents Sl. No. Generic Name Specification Quantity Approx Cost (Rs.) 1. HEPES Molecular biology 1kg 80000/‐ grade 2. MES hydrate Molecular biology 1kg 34000/‐ grade 3. Imidazole Molecular biology 1kg 28000/‐ grade 4. BIS‐TRIS Molecular biology 500g 44000/‐ grade 5. Agarose Type I Molecular biology 1kg 11900/‐ grade 6. CAPS Molecular biology 100g 8000/‐ grade 7. Trizma Base Molecular biology 1kg 31000/‐ grade 8. Glycerol Molecular biology 2.5L*4 8000/‐ grade 9. Ethanol Molecular biology 500ml*10 8000/‐ grade 10. NaCl Molecular biology 1kg 6000/‐ grade 11. Tris Buffer Molecular biology 1 kg 10000/‐ grade 12. Ethyl acetate Molecular biology 500ml*5 6500/‐ grade 13. Acetic acid 25L 11000/‐ 14. Methanol 25L 8000/‐ 15. Urea Molecular biology 1kg 2000/‐ grade 16. Agar Powder, Bacteriological 500g 8000/‐ 17. Trizma Hydrochloride Molecular biology 1kg 15000/‐ grade 18. Acrylamide Molecular biology 500g 4000/‐ grade 19. Phenol Molecular biology 500ml 5000/‐ grade 20. Luria Bertani Broth 2.5kg * 4 90,000 21. Acetone 2.5L 1500/‐ 22. Ni‐NTA beads 100ml 57000/‐ 23. Ammonium persulphate Molecular biology 100g 2000/‐ grade 24. Ampicillin Sodium Molecular biology 100 g 13000/‐ grade 25. Kanaymycin Monosulfate Molecular biology 100g 60000/‐ grade 26. Choramphenicol Molecular biology 100g 40000/‐ grade 27. Isopropyl β‐ d‐1‐thiogalactopyranoside Molecular biology 100g 35000/‐ grade 28. Coomassie Brilliant Blue R‐250 Molecular biology 25g 10,000/‐ grade 29. SDS Molecular biology 100g 2500/‐ grade 30. Isopropanol 25 L 15000/‐ 31. Crystallization screen JCSG‐Plus 1 unit/box 50000/‐ 32. -

Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling
University of Mary Washington Eagle Scholar Student Research Submissions Spring 5-7-2018 Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling Poornima Sunder Follow this and additional works at: https://scholar.umw.edu/student_research Part of the Biochemistry Commons Recommended Citation Sunder, Poornima, "Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling" (2018). Student Research Submissions. 226. https://scholar.umw.edu/student_research/226 This Honors Project is brought to you for free and open access by Eagle Scholar. It has been accepted for inclusion in Student Research Submissions by an authorized administrator of Eagle Scholar. For more information, please contact [email protected]. Synthetic Routes to Bromo-Terminated Phosphonate Films and Alkynyl Pyridine Compounds for Click Coupling Poornima Rachel Sunder Thesis submitted to the faculty of University of Mary Washington in partial fulfillment of the requirements for graduation with Honors in Chemistry (2018) ABSTRACT Click reactions are a highly versatile class of reactions that produce a diverse range of products. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) click reactions require an azide and a terminal alkyne and produce a coupled product that is “clicked” through a triazole ring that can have a variety of substituents. In this work, bromo-terminated phosphonate films on copper oxide surfaces were explored as the platform for click coupling, as the terminal azide needed for the reaction can be generated through an in situ SN2 reaction with a terminal bromo group. The reactions were characterized using model reactions in solution before being conducted on modified copper oxide surfaces. -

Chapter 21 the Chemistry of Carboxylic Acid Derivatives
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 21 The Chemistry of Carboxylic Acid Derivatives Solutions to In-Text Problems 21.1 (b) (d) (e) (h) 21.2 (a) butanenitrile (common: butyronitrile) (c) isopentyl 3-methylbutanoate (common: isoamyl isovalerate) The isoamyl group is the same as an isopentyl or 3-methylbutyl group: (d) N,N-dimethylbenzamide 21.3 The E and Z conformations of N-acetylproline: 21.5 As shown by the data above the problem, a carboxylic acid has a higher boiling point than an ester because it can both donate and accept hydrogen bonds within its liquid state; hydrogen bonding does not occur in the ester. Consequently, pentanoic acid (valeric acid) has a higher boiling point than methyl butanoate. Here are the actual data: INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 21 2 21.7 (a) The carbonyl absorption of the ester occurs at higher frequency, and only the carboxylic acid has the characteristic strong, broad O—H stretching absorption in 2400–3600 cm–1 region. (d) In N-methylpropanamide, the N-methyl group is a doublet at about d 3. N-Ethylacetamide has no doublet resonances. In N-methylpropanamide, the a-protons are a quartet near d 2.5. In N-ethylacetamide, the a- protons are a singlet at d 2. The NMR spectrum of N-methylpropanamide has no singlets. 21.9 (a) The first ester is more basic because its conjugate acid is stabilized not only by resonance interaction with the ester oxygen, but also by resonance interaction with the double bond; that is, the conjugate acid of the first ester has one more important resonance structure than the conjugate acid of the second. -

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

United States Patent 15) 3,669,956 Borck Et Al
United States Patent 15) 3,669,956 Borck et al. (45) June 13, 1972 54) 4-SUBSTITUTEDAMNO 260/472, 260/516, 260/518 R, 260/518 A, 260/519, PHENYACETIC ACDS AND 260/556 AR, 260/556 B, 260/558 S, 260/558 A, DERVATIVES THEREOF 260/559 T, 260/559 A, 260/.571, 260/574, 260/.575, (72 Inventors: Joachim Borck; Johann Dahin; Volker 424/244, 424/246, 424/248, 424/250, 424/267, Koppe; Josef Kramer; Gustav Shorre; J. 424/270, 424/272, 424/273, 424/274, 424/304, W. Hermann Hovy; Ernst Schorscher, all 424/309, 424/32 i, 424/324, 424/330 of Darmstadt, Germany 51) int. Cl. ........................................................ C07d 41/04 58) Field of Search........ 260/294X,293.4, 293.47, 239 BF, 73) Assignee: E. Merck A. G., Darmstadt, Germany 260/326.3, 294.3 E (22) Filed: July 22, 1968 56) References Cited (21) Appl. No.: 746,326 UNITED STATES PATENTS (30) Foreign Application Priority Data 3,252,970 5/1966 Huebner................................ 260/239 July 22, 1967 Germany.............................. M 74881 3,385,852 5/1968 Casadio................................. 260/246 Jan. 8, 1968 Germany... ....M 76850 OTHER PUBLICATIONS Feb. 23, 1968 Germany...... ...M 77363 March 1, 1968 Germany.............................. M 77429 Norman et al., J. Chen. Soc. 1963, (Nov.), 5431-6. (52) U.S. Cl................... 260/239 BF, 260/239 A, 260/239 E, Primary Examiner-Henry R. Jiles 260/243 B, 260/246, 260/247. 1, 260/247.2 R, Assistant Examiner-G. Thomas Todd 260/247.2 A, 260/247.2 B, 260/247.5 R, 260/247.7 Attorney-Millen, Raptes & White A, 260/247.7 H, -

Exam 1 (February 23, 2004) ID# ______
Chemistry 211 Name ___________________________ Exam 1 (February 23, 2004) ID# ___________________________ 10 1. You desire to synthesize 3-ethyl-3-pentanol starting with an ester. (i) What would be the name of the ester, and what is the name for the Grignard reagent (e.g., methyl magnesium bromide)? (ii) For the carbons shown in the product, show plausible hydrocarbons that you could start with to produce the ester and the Grignard reagent (as in a retrosynthesis). 12 2. (i) Show the step-by-step process required to produce propyllithium, which requires a free radical reaction mechanism, . (ii) Show the complete reaction mechanism for reaction between propyllithium and the correct ketone to produce 3-propyl-3-pentanol. (iii) Propose a possible reaction mechanism by which dipropyl cuprate (Cu+ with two propyl groups attached) could react with ethyl bromide to produce a new hydrocarbon. (This is a thinking exercise! So, think! () 8 3. As mentioned in the text, diethyl ether, pentane, and 1-butanol have similar molar masses, but different physical properties. Boiling points are 35oC, 36oC, and 117oC, respectively. Their respective solubilities in water are 7.5g/100mL, insoluble, and 9g/100mL. (i) Draw structures for each of these compounds. (ii) Justify the observed boiling points and their solubilities. 16 4. Draw structures of the following compounds 2,3-heptanediol isopropyllithium benzylmagnesium bromide benzoic acid benzaldehylde dimethyl sulfide t-butyl methanoate dibutyl ketone 12 5. Alcohols can be oxidized to produce other compounds, and can be produced by reduction. For the reactions shown below, show the structure for the expected product (if reaction does not occur, state: No Reaction) when treated with the indicated oxidizing or reducing agents. -

Functionalized Hybrid Silicones – Catalysis, Synthesis and Application
Technische Universität München Fakultät für Chemie Fachgebiet Molekulare Katalyse Functionalized Hybrid Silicones – Catalysis, Synthesis and Application Sophie Luise Miriam Putzien Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. M. Schuster Prüfer der Dissertation: 1. Univ.-Prof. Dr. F. E. Kühn 2. Univ.-Prof. Dr. O.Nuyken (i.R.) Die Dissertation wurde am 16.02.2012 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 08.03.2012 angenommen. The following dissertation was prepared between April 2009 and March 2012 at the Chair of Inorganic Chemistry, Department of Molecular Catalysis of the Technische Universität München. I would like to express my deep gratitude to my academic supervisor Prof. Dr. Fritz E. Kühn for his support and confidence and the freedom of scientific research. This work was supported by a research grant from the BASF Construction Chemicals GmbH, Trostberg, Germany. Acknowledgement I would like to express my sincere gratitude to Prof. Dr. Oskar Nuyken and Dr. Eckhart Louis for their ongoing support and their undamped enthusiasm for my research topic. They supported this work with many inspiring discussions, new ideas and critical questions. I thank the BASF Construction Chemicals GmbH, Trostberg, for giving me the opportunity to work on an industrial cooperation project. Especially, I would like to thank Dr. Simone Klapdohr and Dr. Burkhard Walther, who accompanied this project from the industrial perspectice, for their support and the nice time I had in Trostberg during the application technological tests. -

Chemistry for S5 Students Short Notes & Questions with Answers & And
Chemistry for S5 students Short notes & questions with answers & and other questions to help S5 Students to revise Important Concepts: Chemical Reactions of Alkyl Halides The reaction can be broadly classified in two categories: (a) Nucleophilic substitution (b) Elimination reactions Nucleophilic substitution reactions: In this reaction a nucleophile, which is rich in electrons, attacks partial positive charge on the carbon atom bonded to halogen to replace the leaving group. Nucleophilic reactions proceed by two different mechanism: (a) Substitution nucleophilic bimolecular (SN2) (b) Substitution nucleophilic unirnolecular (SN1) The reaction follows second order kinetics No intermediate is formed. It usually requires a strong nucleophile. The order of reactivity followed as: Primary halide > Secondary halide > Tertiary halide It is carried out in polar protic solvents (water, Alcohol, acetic acid etc.). These reactions occur in two steps as shown above The order of leaving ability is: F- < Cl- < Br- < I- The order of reactivity is as shown below: Difference between E1 and E2 reaction mechanism: Attributes E1 E2 Rate law Depend on the concentration of Depends on the concentration of both substrate substrate and base Barrier Formation of carbocation None 3o>2o>> 1o Base Does not require strong base Requires strong base Stereochemistry Does not require stereochemistry Leaving group must be anti to hydrogen removed Some solved questions are given below: Question 1:Which is the correct increasing order of boiling points of the following compounds? 1-bromoethane, 1-bromopropane, 1-bromobutane, Bromobenzene (a) Bromobenzene < 1-bromobutane < 1-bromopropane < 1-bromoethane (b) Bromobenzene < 1-bromothane < 1-bromopropane < 1-bromobutane (c) 1-bromopropane < 1-bromobutane < 1-bromoethane < Bromobenzene (d) 1-bromoethane < 1-bromopropane < 1-bromobutane < Bromobenzene Solution 1: Boiling point increases with increase in size of hydrocarbon part for the same haloalkanes. -

Bio-Butanol Production from Glycerol with Clostridium
Lin et al. Biotechnol Biofuels (2015) 8:168 DOI 10.1186/s13068-015-0352-6 RESEARCH Open Access Bio‑butanol production from glycerol with Clostridium pasteurianum CH4: the effects of butyrate addition and in situ butanol removal via membrane distillation De‑Shun Lin1, Hong‑Wei Yen2, Wei‑Chen Kao1, Chieh‑Lun Cheng1, Wen‑Ming Chen3, Chieh‑Chen Huang4 and Jo‑Shu Chang1,4,5* Abstract Background: Clostridium pasteurianum CH4 was used to produce butanol from glycerol. The performance of butanol fermentation was improved by adding butyrate as the precursor to trigger the metabolic pathway toward butanol production, and by combining this with in situ butanol removal via vacuum membrane distillation (VMD) to avoid the product inhibition arising from a high butanol concentration. 1 Results: Adding 6 g L− butyrate as precursor led to an increase in the butanol yield from 0.24 to 0.34 mol butanol 1 (mol glycerol)− . Combining VMD and butyrate addition strategies could further enhance the maximum effective 1 1 butanol concentration to 29.8 g L− , while the yield was also improved to 0.39 mol butanol (mol glycerol)− . The butanol concentration in the permeate of VMD was nearly five times higher than that in the feeding solution. Conclusions: The proposed butyrate addition and VMD in situ butanol removal strategies are very effective in enhancing both butanol titer and butanol yield. This would significantly enhance the economic feasibility of fermen‑ tative production of butanol. The VMD-based technology not only alleviates the inhibitory effect of butanol, but also markedly increases butanol concentration in the permeate after condensation, thereby making downstream process‑ ing easier and more cost-effective. -

Enhanced Butanol Production by Free and Immobilized Clostridium Sp
Enhanced Butanol Production by Free and Immobilized Clostridium sp. Cells Using Butyric Acid as Co-Substrate Laili Gholizadeh This thesis comprises 30 ECTS credits and is a compulsory part in the Master of Science with a Major in Chemical Engineering – Applied Biotechnology 120 ECTS credits No. 10/2009 Title: Enhanced Butanol Production by Free and Immobilized Clostridium sp. Cells using Butyric Acid as Co-Substrate. Author: Laili Gholizadeh Baroghi (e-mail: [email protected]) Master Thesis Subject Category: Biotechnology (Bioprocess Engineering – Biofuels) University College of Borås School of Engineering SE-501 90 BORÅS Telephone: (+46) 033 435 4640 Examiner: Prof. Mohammad Taherzadeh Supervisor and Thesis Advisor: Prof. Shang–Tian Yang Supervisor Address: OSU–Ohio State University 125 Koffolt Laboratories 140 West 19th Ave. Columbus, OH 43210–1185, USA Client: Ohio State University (OSU), Chemical & Biomolecular Engineering Department Prof. Shang–Tian Yang Columbus, Ohio; USA. Date: 08–12–2009 Keywords: Bio-butanol y Acetone–Butanol–Ethanol (ABE) y ABE- fermentation y Butyric acid y Clostridium y C. acetobutylicum ATCC 824 y C. beijerinckii ATCC 55025 y C. beijerinckii BA 101 y C. beijerinckii NCIMB 8052 y Fibrous-bed Bioreactor (FBB) y Batch y Suspended cell culture y Immobilized cell system. DEDICATION I would like to dedicate this M.Sc. Thesis to my beloved Family for all their love and encouragement and for always been supportive of my choices. “I am among those who think that science has great beauty. A scientist in his laboratory is not only a technician: he is also a child placed before natural phenomena, which impress him like a fairy tale.” − Marie Curie ABSTRACT Butanol production by four different Clostridium sp. -
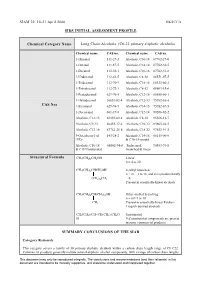
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

The Total Synthesis of Hexavalent Glycodendrimers
THE TOTAL SYNTHESIS OF HEXAVALENT GLYCODENDRIMERS USING A DIVERGENT PATHWAY FOR ANTI-HIV THERAPY A Thesis Presented to the faCulty of the Department of Chemistry California State University, SaCramento Submitted in partial satisfaCtion of the requirements for the degree of MASTER OF SCIENCE in Chemistry by Dustin Andres Dimas SUMMER 2018 © 2018 Dustin Andres Dimas ALL RIGHTS RESERVED ii THE TOTAL SYNTHESIS OF HEXAVALENT GLYCODENDRIMERS USING A DIVERGENT PATHWAY FOR ANTI-HIV THERAPY A Thesis by Dustin Andres Dimas Approved by: __________________________________, Committee Chair Dr. Katherine MCReynolds __________________________________, SeCond Reader Dr. Roy Dixon __________________________________, Third Reader Dr. Cynthia Kellen-Yuen ____________________________ Date iii Student: Dustin Andres Dimas I certify that this student has met the requirements for format contained in the University format manual, and that this thesis is suitable for shelving in the Library and credit is to be awarded for the thesis. __________________________, Graduate Coordinator ___________________ Dr. Susan Crawford Date Department of Chemistry iv AbstraCt of THE TOTAL SYNTHESIS OF HEXAVALENT GLYCODENDRIMERS USING A DIVERGENT PATHWAY FOR ANTI-HIV THERAPY by Dustin Andres Dimas The Human ImmunodefiCiency Virus (HIV) has caused a worldwide epidemiC. Currently, an estimated 36.9 million people are infeCted with HIV. The current treatment for HIV is antiretroviral therapy (ART). Around 20.9 million people living with HIV have aCCess to ART therapy. Right now, only 57% of the people infeCted are currently using ART. As of now, ART can only slow the progression of the virus but doesn’t cure the disease. Additionally, ART can have toxiC side effeCts or beCome ineffeCtive over time through viral resistance.