PDF Available
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Multiyear Survey of Coccidia, Cryptosporidia, Microsporidia, Histomona, and Hematozoa in Wild Quail in the Rolling Plains Ecoregion of Texas and Oklahoma, USA
Journal of Eukaryotic Microbiology ISSN 1066-5234 ORIGINAL ARTICLE Multiyear Survey of Coccidia, Cryptosporidia, Microsporidia, Histomona, and Hematozoa in Wild Quail in the Rolling Plains Ecoregion of Texas and Oklahoma, USA Lixin Xianga,b, Fengguang Guob, Yonglan Yuc, Lacy S. Parsonb, Lloyd LaCosted, Anna Gibsone, Steve M. Presleye, Markus Petersonf, Thomas M. Craigb, Dale Rollinsd,f, Alan M. Fedynichg & Guan Zhub a College of Life Science, Zhejiang University, Hangzhou, Zhejiang 310058, China b Department of Veterinary Pathobiology, College of Veterinary Medicine & Biomedical Sciences, Texas A&M University, College Station, Texas 77843-4467, USA c College of Veterinary Medicine, China Agricultural University, Haidian District, Beijing 100193, China d Rolling Plains Quail Research Foundation, San Angelo, Texas 76901, USA e Institute of Environmental & Human Health, Texas Tech University, Lubbock, Texas 79416, USA f Department of Wildlife & Fisheries Sciences, Texas A&M University, College Station, Texas 77843-2258, USA g Caesar Kleberg Wildlife Research Institute, Texas A&M University-Kingsville, Kingsville, Texas 78363, USA Keywords ABSTRACT Cryptosporidium; molecular epidemiology; northern bobwhite (Colinus virginianus); pro- We developed nested PCR protocols and performed a multiyear survey on the tozoan parasites; scaled quail (Callipepla prevalence of several protozoan parasites in wild northern bobwhite (Colinus squamata). virginianus) and scaled quail (Callipepla squamata) in the Rolling Plains ecore- gion of Texas and Oklahoma (i.e. fecal pellets, bird intestines and blood Correspondence smears collected between 2010 and 2013). Coccidia, cryptosporidia, and G. Zhu, Department of Veterinary Pathobiol- microsporidia were detected in 46.2%, 11.7%, and 44.0% of the samples ogy, College of Veterinary Medicine & (n = 687), whereas histomona and hematozoa were undetected. -

Journal of Parasitology
Journal of Parasitology Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes --Manuscript Draft-- Manuscript Number: 17-111R1 Full Title: Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes Short Title: Eimeria taggarti n. sp. in Prostate of Antechinus flavipes Article Type: Regular Article Corresponding Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc University of Melbourne Melbourne, Victoria AUSTRALIA Corresponding Author Secondary Information: Corresponding Author's Institution: University of Melbourne Corresponding Author's Secondary Institution: First Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc First Author Secondary Information: Order of Authors: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc Joanne Maree Devlin, BVSc(Hons), MVPHMgt, PhD Liliana Tatarczuch David Augustine Taggart David J Schultz Jenny A Charles Ian Beveridge Order of Authors Secondary Information: Abstract: A novel coccidian species was discovered in the prostate of an Antechinus flavipes (yellow-footed antechinus) in South Australia, during the period of post-mating male antechinus immunosuppression and mortality. This novel coccidian is unusual because it develops extra-intestinally and sporulates endogenously within the prostate gland of its mammalian host. Histological examination of prostatic tissue revealed dense aggregations of spherical and thin-walled tetrasporocystic, dizoic sporulated coccidian oocysts within tubular lumina, with unsporulated oocysts and gamogonic stages within the cytoplasm of glandular epithelial cells. This coccidian was observed occurring concurrently with dasyurid herpesvirus 1 infection of the antechinus' prostate. Eimeria- specific 18S small subunit ribosomal DNA PCR amplification was used to obtain a partial 18S rDNA nucleotide sequence from the antechinus coccidian. -

ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central
ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central Texas with an Assessment of Fitness Costs in Erythemis simplicicollis Jason L. Locklin, Ph.D. Mentor: Darrell S. Vodopich, Ph.D. Dragonfly parasites are widespread and frequently include gregarines (Phylum Apicomplexa) in the gut of the host. Gregarines are ubiquitous protozoan parasites that infect arthropods worldwide. More than 1,600 gregarine species have been described, but only a small percentage of invertebrates have been surveyed for these apicomplexan parasites. Some consider gregarines rather harmless, but recent studies suggest otherwise. Odonate-gregarine studies have more commonly involved damselflies, and some have considered gregarines to rarely infect dragonflies. In this study, dragonfly populations were surveyed for gregarines and an assessment of fitness costs was made in a common and widespread host species, Erythemis simplicicollis. Adult dragonfly populations were surveyed weekly at two reservoirs in close proximity to one another and at a flow-through wetland system. Gregarine prevalences and intensities were compared within host populations between genders, among locations, among wing loads, and through time. Host fitness parameters measured included wing load, egg size, clutch size, and total egg count. Of the 37 dragonfly species surveyed, 14 species (38%) hosted gregarines. Thirteen of those species were previously unreported as hosts. Gregarine prevalences ranged from 2% – 52%. Intensities ranged from 1 – 201. Parasites were aggregated among their hosts. Gregarines were found only in individuals exceeding a minimum wing load, indicating that gregarines are likely not transferred from the naiad to adult during emergence. Prevalence and intensity exhibited strong seasonality during both years at one of the reservoirs, but no seasonal trend was detected at the wetland. -
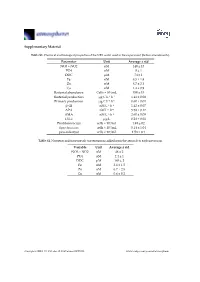
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

A New Species of Hepatozoon (Apicomplexa: Adeleorina) from Python Regius (Serpentes: Pythonidae) and Its Experimental Transmission by a Mosquito Vector
J. Parasitol., 93(?), 2007, pp. 1189–1198 ᭧ American Society of Parasitologists 2007 A NEW SPECIES OF HEPATOZOON (APICOMPLEXA: ADELEORINA) FROM PYTHON REGIUS (SERPENTES: PYTHONIDAE) AND ITS EXPERIMENTAL TRANSMISSION BY A MOSQUITO VECTOR Michal Sloboda, Martin Kamler, Jana Bulantova´*, Jan Voty´pka*†, and David Modry´† Department of Parasitology, University of Veterinary and Pharmaceutical Sciences, Palacke´ho 1-3, 612 42 Brno, Czech Republic. e-mail: [email protected] ABSTRACT: Hepatozoon ayorgbor n. sp. is described from specimens of Python regius imported from Ghana. Gametocytes were found in the peripheral blood of 43 of 55 snakes examined. Localization of gametocytes was mainly inside the erythrocytes; free gametocytes were found in 15 (34.9%) positive specimens. Infections of laboratory-reared Culex quinquefasciatus feeding on infected snakes, as well as experimental infection of juvenile Python regius by ingestion of infected mosquitoes, were performed to complete the life cycle. Similarly, transmission to different snake species (Boa constrictor and Lamprophis fuliginosus) and lizards (Lepidodactylus lugubris) was performed to assess the host specificity. Isolates were compared with Hepatozoon species from sub-Saharan reptiles and described as a new species based on the morphology, phylogenetic analysis, and a complete life cycle. Hemogregarines are the most common intracellular hemo- 3 genera (Telford et al., 2004). Low host specificity of Hepa- parasites found in reptiles. The Hemogregarinidae, Karyolysi- tozoon spp. is supported by experimental transmissions between dae, and Hepatozoidae are distinguished based on the different snakes from different families. Ball (1967) observed experi- developmental patterns in definitive (invertebrate) hosts oper- mental parasitemia with Hepatozoon rarefaciens in the Boa ating as vectors; all 3 families have heteroxenous life cycles constrictor (Boidae); the vector was Culex tarsalis, which had (Telford, 1984). -

Wildlife Parasitology in Australia: Past, Present and Future
CSIRO PUBLISHING Australian Journal of Zoology, 2018, 66, 286–305 Review https://doi.org/10.1071/ZO19017 Wildlife parasitology in Australia: past, present and future David M. Spratt A,C and Ian Beveridge B AAustralian National Wildlife Collection, National Research Collections Australia, CSIRO, GPO Box 1700, Canberra, ACT 2601, Australia. BVeterinary Clinical Centre, Faculty of Veterinary and Agricultural Sciences, University of Melbourne, Werribee, Vic. 3030, Australia. CCorresponding author. Email: [email protected] Abstract. Wildlife parasitology is a highly diverse area of research encompassing many fields including taxonomy, ecology, pathology and epidemiology, and with participants from extremely disparate scientific fields. In addition, the organisms studied are highly dissimilar, ranging from platyhelminths, nematodes and acanthocephalans to insects, arachnids, crustaceans and protists. This review of the parasites of wildlife in Australia highlights the advances made to date, focussing on the work, interests and major findings of researchers over the years and identifies current significant gaps that exist in our understanding. The review is divided into three sections covering protist, helminth and arthropod parasites. The challenge to document the diversity of parasites in Australia continues at a traditional level but the advent of molecular methods has heightened the significance of this issue. Modern methods are providing an avenue for major advances in documenting and restructuring the phylogeny of protistan parasites in particular, while facilitating the recognition of species complexes in helminth taxa previously defined by traditional morphological methods. The life cycles, ecology and general biology of most parasites of wildlife in Australia are extremely poorly understood. While the phylogenetic origins of the Australian vertebrate fauna are complex, so too are the likely origins of their parasites, which do not necessarily mirror those of their hosts. -
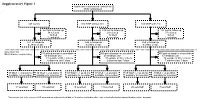
Supplementary Figure 1 Multicenter Randomised Control Trial 2746 Randomised
Supplementary Figure 1 Multicenter randomised control trial 2746 randomised 947 control 910 MNP without zinc 889 MNP with zinc 223 lost to follow up 219 lost to follow up 183 lost to follow up 34 refused 29 refused 37 refused 16 died 12 died 9 died 3 excluded 4 excluded 1 excluded 671 in follow-up 646 in follow-up 659 in follow-up at 24mo of age at 24mo of age at 24mo of age Selection for Microbiome sequencing 516 paired samples unavailable 469 paired samples unavailable 497 paired samples unavailable 69 antibiotic use 63 antibiotic use 67 antibiotic use 31 outside of WLZ criteria 37 outside of WLZ criteria 34 outside of WLZ criteria 6 diarrhea last 7 days 2 diarrhea last 7 days 7 diarrhea last 7 days 39 WLZ > -1 at 24 mo 10 WLZ < -2 at 24mo 58 WLZ > -1 at 24 mo 17 WLZ < -2 at 24mo 48 WLZ > -1 at 24 mo 8 WLZ < -2 at 24mo available for selection available for selection available for selection available for selection available for selection1 available for selection1 14 selected 10 selected 15 selected 14 selected 20 selected1 7 selected1 1 Two subjects (one in the reference WLZ group and one undernourished) had, at 12 months, no diarrhea within 1 day of stool collection but reported diarrhea within 7 days prior. Length, cm kg Weight, Supplementary Figure 2. Length (left) and weight (right) z-scores of children recruited into clinical trial NCT00705445 during the first 24 months of life. Median and quantile values are shown, with medians for participants profiled in current study indicated by red (undernourished) and black (reference WLZ) lines. -

Detection of Cyclospora Cayetanensis, Cryptosporidium Spp., and Toxoplasma Gondii on Imported Leafy Green Vegetables in Canadian Survey
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Food and Waterborne Parasitology 2 (2016) 8–14 Contents lists available at ScienceDirect Food and Waterborne Parasitology journal homepage: www.elsevier.com/locate/fawpar Detection of Cyclospora cayetanensis, Cryptosporidium spp., and Toxoplasma gondii on imported leafy green vegetables in Canadian survey Laura F. Lalonde, Alvin A. Gajadhar ⁎ Centre for Food-borne and Animal Parasitology, Canadian Food Inspection Agency, Saskatoon Laboratory, 116 Veterinary Road, Saskatoon, Saskatchewan S7N 2R3, Canada article info abstract Article history: A national survey was performed to determine the prevalence of Cyclospora cayetanensis, Received 17 November 2015 Cryptosporidium spp., and Toxoplasma gondii in leafy green vegetables (leafy greens) purchased Received in revised form 29 January 2016 at retail in Canada. A total of 1171 samples of pre-packaged or bulk leafy greens from domestic Accepted 29 January 2016 (24.25%) and imported (75.75%) sources were collected at retail outlets from 11 Canadian cities Available online 23 February 2016 between April 2014 and March 2015. The samples were processed by shaking or stomaching in an elution buffer followed by oocyst isolation and concentration. DNA extracted from the wash Keywords: concentrates was tested for C. cayetanensis, Cryptosporidium spp., and T. gondii using our previ- Leafy green vegetables ously developed and validated 18S rDNA qPCR assay with a universal coccidia primer cocktail Food safety and melting curve analysis. Test samples that amplified and had a melting temperature and Cyclospora Cryptosporidium melt curve shape matching the C. cayetanensis, C. parvum, C. -

Protistology an International Journal Vol
Protistology An International Journal Vol. 10, Number 2, 2016 ___________________________________________________________________________________ CONTENTS INTERNATIONAL SCIENTIFIC FORUM «PROTIST–2016» Yuri Mazei (Vice-Chairman) Welcome Address 2 Organizing Committee 3 Organizers and Sponsors 4 Abstracts 5 Author Index 94 Forum “PROTIST-2016” June 6–10, 2016 Moscow, Russia Website: http://onlinereg.ru/protist-2016 WELCOME ADDRESS Dear colleagues! Republic) entitled “Diplonemids – new kids on the block”. The third lecture will be given by Alexey The Forum “PROTIST–2016” aims at gathering Smirnov (Saint Petersburg State University, Russia): the researchers in all protistological fields, from “Phylogeny, diversity, and evolution of Amoebozoa: molecular biology to ecology, to stimulate cross- new findings and new problems”. Then Sandra disciplinary interactions and establish long-term Baldauf (Uppsala University, Sweden) will make a international scientific cooperation. The conference plenary presentation “The search for the eukaryote will cover a wide range of fundamental and applied root, now you see it now you don’t”, and the fifth topics in Protistology, with the major focus on plenary lecture “Protist-based methods for assessing evolution and phylogeny, taxonomy, systematics and marine water quality” will be made by Alan Warren DNA barcoding, genomics and molecular biology, (Natural History Museum, United Kingdom). cell biology, organismal biology, parasitology, diversity and biogeography, ecology of soil and There will be two symposia sponsored by ISoP: aquatic protists, bioindicators and palaeoecology. “Integrative co-evolution between mitochondria and their hosts” organized by Sergio A. Muñoz- The Forum is organized jointly by the International Gómez, Claudio H. Slamovits, and Andrew J. Society of Protistologists (ISoP), International Roger, and “Protists of Marine Sediments” orga- Society for Evolutionary Protistology (ISEP), nized by Jun Gong and Virginia Edgcomb. -

Haemocystidium Spp., a Species Complex Infecting Ancient Aquatic Turtles of the Family Podocnemididae First Report of These
IJP: Parasites and Wildlife 10 (2019) 299–309 Contents lists available at ScienceDirect IJP: Parasites and Wildlife journal homepage: www.elsevier.com/locate/ijppaw Haemocystidium spp., a species complex infecting ancient aquatic turtles of the family Podocnemididae: First report of these parasites in Podocnemis T vogli from the Orinoquia Leydy P. Gonzáleza,b, M. Andreína Pachecoc, Ananías A. Escalantec, Andrés David Jiménez Maldonadoa,d, Axl S. Cepedaa, Oscar A. Rodríguez-Fandiñoe, ∗ Mario Vargas‐Ramírezd, Nubia E. Mattaa, a Departamento de Biología, Facultad de Ciencias, Universidad Nacional de Colombia, Sede Bogotá, Carrera 30 No 45-03, Bogotá, Colombia b Instituto de Biotecnología, Facultad de Ciencias, Universidad Nacional de Colombia, Sede Bogotá, Carrera 30 No 45-03, Bogotá, Colombia c Department of Biology/Institute for Genomics and Evolutionary Medicine (iGEM), Temple University, Philadelphia, PA, USA d Instituto de Genética, Universidad Nacional de Colombia, Sede Bogotá, Carrera 30 No 45-03, Bogotá, Colombia e Fundación Universitaria-Unitrópico, Dirección de Investigación, Grupo de Investigación en Ciencias Biológicas de la Orinoquía (GINBIO), Colombia ARTICLE INFO ABSTRACT Keywords: The genus Haemocystidium was described in 1904 by Castellani and Willey. However, several studies considered Haemoparasites it a synonym of the genera Plasmodium or Haemoproteus. Recently, molecular evidence has shown the existence Reptile of a monophyletic group that corresponds to the genus Haemocystidium. Here, we further explore the clade Simondia Haemocystidium spp. by studying parasites from Testudines. A total of 193 individuals belonging to six families of Chelonians Testudines were analyzed. The samples were collected in five localities in Colombia: Casanare, Vichada, Arauca, Colombia Antioquia, and Córdoba. From each individual, a blood sample was taken for molecular analysis, and peripheral blood smears were made, which were fixed and subsequently stained with Giemsa. -

A MOLECULAR PHYLOGENY of MALARIAL PARASITES RECOVERED from CYTOCHROME B GENE SEQUENCES
J. Parasitol., 88(5), 2002, pp. 972±978 q American Society of Parasitologists 2002 A MOLECULAR PHYLOGENY OF MALARIAL PARASITES RECOVERED FROM CYTOCHROME b GENE SEQUENCES Susan L. Perkins* and Jos. J. Schall Department of Biology, University of Vermont, Burlington, Vermont 05405. e-mail: [email protected] ABSTRACT: A phylogeny of haemosporidian parasites (phylum Apicomplexa, family Plasmodiidae) was recovered using mito- chondrial cytochrome b gene sequences from 52 species in 4 genera (Plasmodium, Hepatocystis, Haemoproteus, and Leucocy- tozoon), including parasite species infecting mammals, birds, and reptiles from over a wide geographic range. Leucocytozoon species emerged as an appropriate out-group for the other malarial parasites. Both parsimony and maximum-likelihood analyses produced similar phylogenetic trees. Life-history traits and parasite morphology, traditionally used as taxonomic characters, are largely phylogenetically uninformative. The Plasmodium and Hepatocystis species of mammalian hosts form 1 well-supported clade, and the Plasmodium and Haemoproteus species of birds and lizards form a second. Within this second clade, the relation- ships between taxa are more complex. Although jackknife support is weak, the Plasmodium of birds may form 1 clade and the Haemoproteus of birds another clade, but the parasites of lizards fall into several clusters, suggesting a more ancient and complex evolutionary history. The parasites currently placed within the genus Haemoproteus may not be monophyletic. Plasmodium falciparum of humans was not derived from an avian malarial ancestor and, except for its close sister species, P. reichenowi,is only distantly related to haemospordian parasites of all other mammals. Plasmodium is paraphyletic with respect to 2 other genera of malarial parasites, Haemoproteus and Hepatocystis. -

Download Article (PDF)
OCCASONAL PAPER NO. 48 RECORDS OF THE ZOOLOGICAL SURVEY OF INDIA MISCELLANEOUS PUBLICATION OCCASIONAL PAPER NO. 48 INDEX-CATALOGUE OF AVIAN HAEMATOZOA FROM INDIA By N. C. NANDI Zoological Survey of India, Kakdwip Field Station Kakd\vip, West Bengal ~ar1fI Edited by the Director, Zoological Survey of India 1984 C Copyright, Government of India, 1984 Published in May, 1984 PRICE: Inland: Rs. 27-00 Foreign: £ 3-00 S 6-00 PRINTED IN INDIA AT IMPRINTA, 243/2B, A. P. c. ROAD, CALCUTTA-6 AND PUBLISHED BY THB DIRECTOR, ZOOLOGICAL SURVEY OF INDIA, CALCUTrA RECORDS OF THE ZOOLOGICAL SURVEY OF INDIA MISCELLANEOUS PUBLICATION OCCASIONAL PAPER No. 48 1984 Pages 1-64 CONTENTS Pages INTRODUCTION ... 1 SYSTEMATIC POSITION OF THB AVIAN HABMATOZOA 1 SPECIES CATALOGUE 3 Genus Trypanosoma 3 Lankesterella 5 Toxoplasma 5 LeucocytozoOD 6 Haemoproteus 8 Plasmodium 15 Babesia 17 M i crofila ria 17 Unidentified parasites 20 HOST CATALOGUE 21 Order Ciconiformes 21 Family Ardeidae 21 Family Threskiornithidae .00 21 Order Anseriformes 21 Family Anatidae ... ... 21 Order Falconiformes ... ... 21 Family Accipitridae 00' 21 Family Falconidae 22 Order Galliformes .. , 22 Family Megapodiidae 22 Family Phasianidae .0' 22 Order Gruiformes .00 23 Family Turnicidae 23 Family Rallidae ... 23 Order Charadriiformes ." 24 Family Charadriidae 24 Order Columbiformes 24 Family Columbidae 24 ( ii ) Order Psittaciformes 25 Family Psittacidae ... 25 Order Cuculiformes 2S Family Cuculidae 25 Order Strigiformes 25 Family Strigidae 25 Order Coraciiformes 26 Family Alcedinidae 26 Family Meropidae 26 Family Coraciidae 27 Family Upupidae 27 Order Piciformes 27 Family Capitonidae 27 Family Picidae 28 Order Passeriformes 28 Family Pittidae ... 28 Alaudidae ... 29 Campephagidae 29 Dicruridae 29 Oriolidae 29 Corvidae 30 Paridae 31 Sittidae 31 Timaliidae 3L Pycnonotidae 32 Aegithinidae 33 Turdidae 33 Sylviidae 34 M uscicapidae 35 Motacillidae 36 Laniidae ..