Metabarcoding Analysis of Prey Composition of the Copepod Calanus Finmarchicus in Regions of the North Atlantic Ocean Heidi Yeh [email protected]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Molecular Data and the Evolutionary History of Dinoflagellates by Juan Fernando Saldarriaga Echavarria Diplom, Ruprecht-Karls-Un
Molecular data and the evolutionary history of dinoflagellates by Juan Fernando Saldarriaga Echavarria Diplom, Ruprecht-Karls-Universitat Heidelberg, 1993 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE STUDIES Department of Botany We accept this thesis as conforming to the required standard THE UNIVERSITY OF BRITISH COLUMBIA November 2003 © Juan Fernando Saldarriaga Echavarria, 2003 ABSTRACT New sequences of ribosomal and protein genes were combined with available morphological and paleontological data to produce a phylogenetic framework for dinoflagellates. The evolutionary history of some of the major morphological features of the group was then investigated in the light of that framework. Phylogenetic trees of dinoflagellates based on the small subunit ribosomal RNA gene (SSU) are generally poorly resolved but include many well- supported clades, and while combined analyses of SSU and LSU (large subunit ribosomal RNA) improve the support for several nodes, they are still generally unsatisfactory. Protein-gene based trees lack the degree of species representation necessary for meaningful in-group phylogenetic analyses, but do provide important insights to the phylogenetic position of dinoflagellates as a whole and on the identity of their close relatives. Molecular data agree with paleontology in suggesting an early evolutionary radiation of the group, but whereas paleontological data include only taxa with fossilizable cysts, the new data examined here establish that this radiation event included all dinokaryotic lineages, including athecate forms. Plastids were lost and replaced many times in dinoflagellates, a situation entirely unique for this group. Histones could well have been lost earlier in the lineage than previously assumed. -

Growth and Grazing Rates of the Herbivorous Dinoflagellate Gymnodinium Sp
MARINE ECOLOGY PROGRESS SERIES Published December 16 Mar. Ecol. Prog. Ser. Growth and grazing rates of the herbivorous dinoflagellate Gymnodinium sp. from the open subarctic Pacific Ocean Suzanne L. Strom' School of Oceanography WB-10, University of Washington. Seattle. Washington 98195, USA ABSTRACT: Growth, grazing and cell volume of the small heterotroph~cdinoflagellate Gyrnnodin~um sp. Isolated from the open subarctic Pacific Ocean were measured as a funct~onof food concentration using 2 phytoplankton food species. Growth and lngestlon rates increased asymptotically with Increas- ing phytoplankon food levels, as did grazer cell volume; rates at representative oceanic food levels were high but below maxima. Clearance rates decreased with lncreaslng food levels when Isochrysis galbana was the food source; they increased ~vithlncreaslng food levels when Synechococcus sp. was the food source. There was apparently a grazlng threshold for Ingestion of Synechococcus: below an initial Synechococcus concentration of 20 pgC 1.' ingestion rates on this alga were very low, while above this initial concentratlon Synechococcus was grazed preferent~ally Gross growth efficiency varied between 0.03 and 0.53 (mean 0.21) and was highest at low food concentrations. Results support the hypothesis that heterotrophic d~noflagellatesmay contribute to controlling population increases of small, rap~dly-grow~ngphytoplankton specles even at low oceanic phytoplankton concentrations. INTRODUCTION as Gymnodinium and Gyrodinium is difficult or impos- sible using older preservation and microscopy tech- Heterotrophic dinoflagellates can be a significant niques; experimental emphasis has been on more component of the microzooplankton in marine waters. easily recognizable and collectable microzooplankton In the oceanic realm, Lessard (1984) and Shapiro et al. -
![28-Protistsf20r.Ppt [Compatibility Mode]](https://docslib.b-cdn.net/cover/9929/28-protistsf20r-ppt-compatibility-mode-159929.webp)
28-Protistsf20r.Ppt [Compatibility Mode]
9/3/20 Ch 28: The Protists (a.k.a. Protoctists) (meet these in more detail in your book and lab) 1 Protists invent: eukaryotic cells size complexity Remember: 1°(primary) endosymbiosis? -> mitochondrion -> chloroplast genome unicellular -> multicellular 2 1 9/3/20 For chloroplasts 2° (secondary) happened (more complicated) {3°(tertiary) happened too} 3 4 Eukaryotic “supergroups” (SG; between K and P) 4 2 9/3/20 Protists invent sex: meiosis and fertilization -> 3 Life Cycles/Histories (Fig 13.6) Spores and some protists (Humans do this one) 5 “Algae” Group PS Pigments Euglenoids chl a & b (& carotenoids) Dinoflagellates chl a & c (usually) (& carotenoids) Diatoms chl a & c (& carotenoids) Xanthophytes chl a & c (& carotenoids) Chrysophytes chl a & c (& carotenoids) Coccolithophorids chl a & c (& carotenoids) Browns chl a & c (& carotenoids) Reds chl a, phycobilins (& carotenoids) Greens chl a & b (& carotenoids) (more groups exist) 6 3 9/3/20 Name word roots (indicate nutrition) “algae” (-phyt-) protozoa (no consistent word ending) “fungal-like” (-myc-) Ecological terms plankton phytoplankton zooplankton 7 SG: Excavata/Excavates “excavated” feeding groove some have reduced mitochondria (e.g.: mitosomes, hydrogenosomes) 8 4 9/3/20 SG: Excavata O: Diplomonads: †Giardia Cl: Parabasalids: Trichonympha (bk only) †Trichomonas P: Euglenophyta/zoa C: Kinetoplastids = trypanosomes/hemoflagellates: †Trypanosoma C: Euglenids: Euglena 9 SG: “SAR” clade: Clade Alveolates cell membrane 10 5 9/3/20 SG: “SAR” clade: Clade Alveolates P: Dinoflagellata/Pyrrophyta: -

The Planktonic Protist Interactome: Where Do We Stand After a Century of Research?
bioRxiv preprint doi: https://doi.org/10.1101/587352; this version posted May 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Bjorbækmo et al., 23.03.2019 – preprint copy - BioRxiv The planktonic protist interactome: where do we stand after a century of research? Marit F. Markussen Bjorbækmo1*, Andreas Evenstad1* and Line Lieblein Røsæg1*, Anders K. Krabberød1**, and Ramiro Logares2,1** 1 University of Oslo, Department of Biosciences, Section for Genetics and Evolutionary Biology (Evogene), Blindernv. 31, N- 0316 Oslo, Norway 2 Institut de Ciències del Mar (CSIC), Passeig Marítim de la Barceloneta, 37-49, ES-08003, Barcelona, Catalonia, Spain * The three authors contributed equally ** Corresponding authors: Ramiro Logares: Institute of Marine Sciences (ICM-CSIC), Passeig Marítim de la Barceloneta 37-49, 08003, Barcelona, Catalonia, Spain. Phone: 34-93-2309500; Fax: 34-93-2309555. [email protected] Anders K. Krabberød: University of Oslo, Department of Biosciences, Section for Genetics and Evolutionary Biology (Evogene), Blindernv. 31, N-0316 Oslo, Norway. Phone +47 22845986, Fax: +47 22854726. [email protected] Abstract Microbial interactions are crucial for Earth ecosystem function, yet our knowledge about them is limited and has so far mainly existed as scattered records. Here, we have surveyed the literature involving planktonic protist interactions and gathered the information in a manually curated Protist Interaction DAtabase (PIDA). In total, we have registered ~2,500 ecological interactions from ~500 publications, spanning the last 150 years. -

ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central
ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central Texas with an Assessment of Fitness Costs in Erythemis simplicicollis Jason L. Locklin, Ph.D. Mentor: Darrell S. Vodopich, Ph.D. Dragonfly parasites are widespread and frequently include gregarines (Phylum Apicomplexa) in the gut of the host. Gregarines are ubiquitous protozoan parasites that infect arthropods worldwide. More than 1,600 gregarine species have been described, but only a small percentage of invertebrates have been surveyed for these apicomplexan parasites. Some consider gregarines rather harmless, but recent studies suggest otherwise. Odonate-gregarine studies have more commonly involved damselflies, and some have considered gregarines to rarely infect dragonflies. In this study, dragonfly populations were surveyed for gregarines and an assessment of fitness costs was made in a common and widespread host species, Erythemis simplicicollis. Adult dragonfly populations were surveyed weekly at two reservoirs in close proximity to one another and at a flow-through wetland system. Gregarine prevalences and intensities were compared within host populations between genders, among locations, among wing loads, and through time. Host fitness parameters measured included wing load, egg size, clutch size, and total egg count. Of the 37 dragonfly species surveyed, 14 species (38%) hosted gregarines. Thirteen of those species were previously unreported as hosts. Gregarine prevalences ranged from 2% – 52%. Intensities ranged from 1 – 201. Parasites were aggregated among their hosts. Gregarines were found only in individuals exceeding a minimum wing load, indicating that gregarines are likely not transferred from the naiad to adult during emergence. Prevalence and intensity exhibited strong seasonality during both years at one of the reservoirs, but no seasonal trend was detected at the wetland. -

Grazing Impacts of the Heterotrophic Dinoflagellate Polykrikos Kofoidii on a Bloom of Gymnodinium Catenatum
AQUATIC MICROBIAL ECOLOGY Published April 30 Aquat Microb Ecol NOTE Grazing impacts of the heterotrophic dinoflagellate Polykrikos kofoidii on a bloom of Gymnodinium catenatum Yukihiko Matsuyama'f*,Masahide Miyamoto2, Yuichi ~otani' 'National Research Institute of Fisheries and Environment of Inland Sea, Maruishi, Ohno, Saeki, Hiroshima 739-0452, Japan 2KumamotoAriake Fisheries Direction Office, Iwasaki, Tamana, Kumamoto 865-0016, Japan ABSTRACT: In 1998, a red tide of the paralytic shellfish an assessment of the natural population of G. catena- poisoning (PSP)-producing dinoflagellate Gymnodinium cate- turn coupled with a laboratory incubation experiment naturn Graham occurred in Yatsushiro Sea, western Japan. to evaluate the bloom fate. We present data showing The dramatic decline of dominant G. catenatum cells oc- curred during the field and laboratory assessments, accompa- considerable predation by the pseudocolonial hetero- nied with growth of the heterotrophic dinoflagellate Poly- trophic dinoflagellate Polykrikos kofoidii Chatton on knkos kofoidii Chatton. Microscopic observations on both the dominant G. catenatum population, and discuss field and laboratory cultured bloom water revealed that the ecological importance of the genus Polykrikos and >50% of P. kofoidii predated on the natural population of G. catenaturn, and 1 to 8 G. catenatum cells were found in its grazing impact on harmful algal blooms. food vacuoles of P. kofoidii pseudocolonies. Our results sug- Materials and methods. Filed population surveys: gest that predation by P. kofoidii contributes to the cessation The Gymnodinium catenatum bloom occurred from 19 of a G. catenatum bloom. January to 5 February in Miyano-Gawachi Bay, west- ern Yatsushiro Sea, Kyushu Island (Fig. 1). Five cruises KEY WORDS: PSP - Gymnodimurn catenatum . -

A Parasite of Marine Rotifers: a New Lineage of Dinokaryotic Dinoflagellates (Dinophyceae)
Hindawi Publishing Corporation Journal of Marine Biology Volume 2015, Article ID 614609, 5 pages http://dx.doi.org/10.1155/2015/614609 Research Article A Parasite of Marine Rotifers: A New Lineage of Dinokaryotic Dinoflagellates (Dinophyceae) Fernando Gómez1 and Alf Skovgaard2 1 Laboratory of Plankton Systems, Oceanographic Institute, University of Sao˜ Paulo, Prac¸a do Oceanografico´ 191, Cidade Universitaria,´ 05508-900 Butanta,˜ SP, Brazil 2Department of Veterinary Disease Biology, University of Copenhagen, Stigbøjlen 7, 1870 Frederiksberg C, Denmark Correspondence should be addressed to Fernando Gomez;´ [email protected] Received 11 July 2015; Accepted 27 August 2015 Academic Editor: Gerardo R. Vasta Copyright © 2015 F. Gomez´ and A. Skovgaard. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Dinoflagellate infections have been reported for different protistan and animal hosts. We report, for the first time, the association between a dinoflagellate parasite and a rotifer host, tentatively Synchaeta sp. (Rotifera), collected from the port of Valencia, NW Mediterranean Sea. The rotifer contained a sporangium with 100–200 thecate dinospores that develop synchronically through palintomic sporogenesis. This undescribed dinoflagellate forms a new and divergent fast-evolved lineage that branches amongthe dinokaryotic dinoflagellates. 1. Introduction form independent lineages with no evident relation to other dinoflagellates [12]. In this study, we describe a new lineage of The alveolates (or Alveolata) are a major lineage of protists an undescribed parasitic dinoflagellate that largely diverged divided into three main phyla: ciliates, apicomplexans, and from other known dinoflagellates. -

Metagenomic Characterization of Unicellular Eukaryotes in the Urban Thessaloniki Bay
Metagenomic characterization of unicellular eukaryotes in the urban Thessaloniki Bay George Tsipas SCHOOL OF ECONOMICS, BUSINESS ADMINISTRATION & LEGAL STUDIES A thesis submitted for the degree of Master of Science (MSc) in Bioeconomy Law, Regulation and Management May, 2019 Thessaloniki – Greece George Tsipas ’’Metagenomic characterization of unicellular eukaryotes in the urban Thessaloniki Bay’’ Student Name: George Tsipas SID: 268186037282 Supervisor: Prof. Dr. Savvas Genitsaris I hereby declare that the work submitted is mine and that where I have made use of another’s work, I have attributed the source(s) according to the Regulations set in the Student’s Handbook. May, 2019 Thessaloniki - Greece Page 2 of 63 George Tsipas ’’Metagenomic characterization of unicellular eukaryotes in the urban Thessaloniki Bay’’ 1. Abstract The present research investigates through metagenomics sequencing the unicellular protistan communities in Thermaikos Gulf. This research analyzes the diversity, composition and abundance in this marine environment. Water samples were collected monthly from April 2017 to February 2018 in the port of Thessaloniki (Harbor site, 40o 37’ 55 N, 22o 56’ 09 E). The extraction of DNA was completed as well as the sequencing was performed, before the downstream read processing and the taxonomic classification that was assigned using PR2 database. A total of 1248 Operational Taxonomic Units (OTUs) were detected but only 700 unicellular eukaryotes were analyzed, excluding unclassified OTUs, Metazoa and Streptophyta. In this research-based study the most abundant and diverse taxonomic groups were Dinoflagellata and Protalveolata. Specifically, the most abundant groups of all samples are Dinoflagellata with 190 OTUs (27.70%), Protalveolata with 139 OTUs (20.26%) Ochrophyta with 73 OTUs (10.64%), Cercozoa with 67 OTUs (9.77%) and Ciliophora with 64 OTUs (9.33%). -
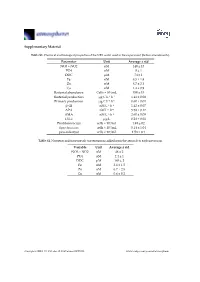
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

Predatory Flagellates – the New Recently Discovered Deep Branches of the Eukaryotic Tree and Their Evolutionary and Ecological Significance
Protistology 14 (1), 15–22 (2020) Protistology Predatory flagellates – the new recently discovered deep branches of the eukaryotic tree and their evolutionary and ecological significance Denis V. Tikhonenkov Papanin Institute for Biology of Inland Waters, Russian Academy of Sciences, Borok, 152742, Russia | Submitted March 20, 2020 | Accepted April 6, 2020 | Summary Predatory protists are poorly studied, although they are often representing important deep-branching evolutionary lineages and new eukaryotic supergroups. This short review/opinion paper is inspired by the recent discoveries of various predatory flagellates, which form sister groups of the giant eukaryotic clusters on phylogenetic trees, and illustrate an ancestral state of one or another supergroup of eukaryotes. Here we discuss their evolutionary and ecological relevance and show that the study of such protists may be essential in addressing previously puzzling evolutionary problems, such as the origin of multicellular animals, the plastid spread trajectory, origins of photosynthesis and parasitism, evolution of mitochondrial genomes. Key words: evolution of eukaryotes, heterotrophic flagellates, mitochondrial genome, origin of animals, photosynthesis, predatory protists, tree of life Predatory flagellates and diversity of eu- of the hidden diversity of protists (Moon-van der karyotes Staay et al., 2000; López-García et al., 2001; Edg- comb et al., 2002; Massana et al., 2004; Richards The well-studied multicellular animals, plants and Bass, 2005; Tarbe et al., 2011; de Vargas et al., and fungi immediately come to mind when we hear 2015). In particular, several prevailing and very abun- the term “eukaryotes”. However, these groups of dant ribogroups such as MALV, MAST, MAOP, organisms represent a minority in the real diversity MAFO (marine alveolates, stramenopiles, opistho- of evolutionary lineages of eukaryotes. -

Bioluminescence As an Ecological Factor During High Arctic Polar Night Heather A
www.nature.com/scientificreports OPEN Bioluminescence as an ecological factor during high Arctic polar night Heather A. Cronin1, Jonathan H. Cohen1, Jørgen Berge2,3, Geir Johnsen3,4 & Mark A. Moline1 Bioluminescence commonly infuences pelagic trophic interactions at mesopelagic depths. Here receie: 01 pri 016 we characterize a vertical gradient in structure of a generally low species diversity bioluminescent ccepte: 14 Octoer 016 community at shallower epipelagic depths during the polar night period in a high Arctic ford with in Puise: 0 oemer 016 situ bathyphotometric sampling. Bioluminescence potential of the community increased with depth to a peak at 80 m. Community composition changed over this range, with an ecotone at 20–40 m where a dinofagellate-dominated community transitioned to dominance by the copepod Metridia longa. Coincident at this depth was bioluminescence exceeding atmospheric light in the ambient pelagic photon budget, which we term the bioluminescence compensation depth. Collectively, we show a winter bioluminescent community in the high Arctic with vertical structure linked to attenuation of atmospheric light, which has the potential to infuence pelagic ecology during the light-limited polar night. Light and vision play a large role in interactions among organisms in both the epipelagic (0–200 m) and mesope- lagic (200–1000 m) realms1,2. Eye structure and function in these habitats is commonly adapted for photon capture in the underwater light feld, with increasing specialization in the mesopelagic3. To avoid visual detection, species in epi- and mesopelagic habitats employ cryptic strategies such as transparency4 and counter-illumination5,6, along with diel vertical migration7,8, to remain hidden from potential predators. -
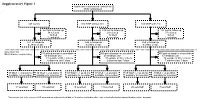
Supplementary Figure 1 Multicenter Randomised Control Trial 2746 Randomised
Supplementary Figure 1 Multicenter randomised control trial 2746 randomised 947 control 910 MNP without zinc 889 MNP with zinc 223 lost to follow up 219 lost to follow up 183 lost to follow up 34 refused 29 refused 37 refused 16 died 12 died 9 died 3 excluded 4 excluded 1 excluded 671 in follow-up 646 in follow-up 659 in follow-up at 24mo of age at 24mo of age at 24mo of age Selection for Microbiome sequencing 516 paired samples unavailable 469 paired samples unavailable 497 paired samples unavailable 69 antibiotic use 63 antibiotic use 67 antibiotic use 31 outside of WLZ criteria 37 outside of WLZ criteria 34 outside of WLZ criteria 6 diarrhea last 7 days 2 diarrhea last 7 days 7 diarrhea last 7 days 39 WLZ > -1 at 24 mo 10 WLZ < -2 at 24mo 58 WLZ > -1 at 24 mo 17 WLZ < -2 at 24mo 48 WLZ > -1 at 24 mo 8 WLZ < -2 at 24mo available for selection available for selection available for selection available for selection available for selection1 available for selection1 14 selected 10 selected 15 selected 14 selected 20 selected1 7 selected1 1 Two subjects (one in the reference WLZ group and one undernourished) had, at 12 months, no diarrhea within 1 day of stool collection but reported diarrhea within 7 days prior. Length, cm kg Weight, Supplementary Figure 2. Length (left) and weight (right) z-scores of children recruited into clinical trial NCT00705445 during the first 24 months of life. Median and quantile values are shown, with medians for participants profiled in current study indicated by red (undernourished) and black (reference WLZ) lines.