A Soft-Chemistry Approach to the Synthesis of Amorphous Calcium Ortho/Pyrophosphate Biomaterials of Tunable Composition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
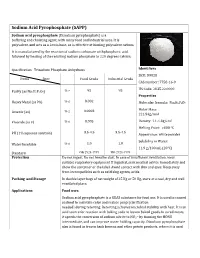
Sodium Acid Pyrophosphate (SAPP)
Sodium Acid Pyrophosphate (SAPP) Sodium acid pyrophosphate (Disodium pyrophosphate) is a buffering and chelating agent, with many food and industrial uses. It is polyvalent, and acts as a Lewis base, so is effective at binding polyvalent cations. It is manufactured by the reaction of sodium carbonate withphosphoric acid. followed by heating of the resulting sodium phosphate to 220 degrees Celsius. Specification: Trisodium Phosphate Anhydrous Identifiers SKU: D9028 Items Spec Food Grade Industrial Grade CAS number: 7758-16-9 HS Code: 2835.22.0000 Purity (as Na2H2P2O7) % ≥ 95 95 Properties % ≤ 0.002 Heavy Metal (as Pb) Molecular formula: Na2H2P2O7 Molar Mass: Arsenic (as) % ≤ 0.0003 221.94g/mol Fluoride (as F) % ≤ 0.005 Density: 1.1-1.3g/cm3 Melting Point: >600 °C 3.5-4.5 3.5-4.5 PH (1% aqueous solution) Appearance: white powder Solubility in Water: Water Insoluble % ≤ 1.0 1.0 11.9 g/100 mL (20°C ) Standard GB 2928-1999 HG 2928-1999 Protection Do not ingest. Do not breathe dust. In case of insufficient ventilation, wear suitable respiratory equipment If ingested, seek medical advice immediately and show the container or the label. Avoid contact with skin and eyes. Keep away from incompatibles such as oxidizing agents, acids. Packing and Storage In double layer bags of net weight of 25 Kg or 50 Kg, store at a cool, dry and well ventilated place. Applications Food uses Sodium acid pyrophosphate is a GRAS substance for food use. It is used in canned seafood to maintain color and reduce purge[clarification needed] during retorting. Retorting achieves microbial stability with heat. -

List of Designated Food Additives
The Japan Food chemical Research Faundation List of Designated Food Additives The substances below are the designated food additives appearing in Appended Table 1, as mentioned in Article 12 of the Enforcement Regulations under the Food Sanitation Law. These additives are listed here in alphabetical order. They are 472 in total as of January 15, 2021. The number preceding the name of each additive is the sequence number given to the corresponding additive in the original Japanese table.The names of the substances are translated by the Foundation from the Japanese original. 16 Acesulfame Potassium(Acesulfame K) 20 Acetaldehyde 332 Glacial Acetic Acid 23 Acetone 22 Acetophenone 17 Acetylated Distarch Adipate 19 Acetylated Distarch Phosphate 18 Acetylated Oxidized Starch 5 Adipic Acid 26 Advantame 32 DL-Alanine 196 Aliphatic Higher Alcohols 197 Aliphatic Higher Aldehydes (except those generally recognized as highly toxic) 198 Aliphatic Higher Hydrocarbons (except those generally recognized as highly toxic) 183 Allyl Cyclohexylpropionate 377 Allyl Hexanoate (Allyl Caproate) 55 Allyl Isothiocyanate (Volatile Oil of Mustard) 446 Aluminum Ammonium Sulfate (Crystal: Ammonium Alum, Dried: Burnt Ammonium Alum) 447 Aluminum Potassium Sulfate (Crystal: Alum or Potassium Alum, Dried: Burnt Alum) 29 (3-Amino-3-carboxypropyl)dimethylsulfonium Chloride 45 Ammonia 36 Ammonium Alginate 248 Ammonium Bicarbonate (Ammonium Hydrogen Carbonate) 245 Ammonium Carbonate 85 Ammonium Chloride 463 Ammonium Dihydrogen Phosphate (Ammonium Phosphate, Monobasic) 33 Ammonium -
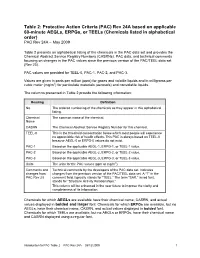
Rev. 24A Table 2: Pacs by Chemical Name
Table 2: Protective Action Criteria (PAC) Rev 24A based on applicable 60-minute AEGLs, ERPGs, or TEELs (Chemicals listed in alphabetical order) PAC Rev 24A – May 2009 Table 2 presents an alphabetical listing of the chemicals in the PAC data set and provides the Chemical Abstract Service Registry Numbers (CASRNs), PAC data, and technical comments focusing on changes in the PAC values since the previous version of the PAC/TEEL data set (Rev 23). PAC values are provided for TEEL-0, PAC-1, PAC-2, and PAC-3. Values are given in parts per million (ppm) for gases and volatile liquids and in milligrams per cubic meter (mg/m3) for particulate materials (aerosols) and nonvolatile liquids. The columns presented in Table 2 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing. Chemical The common name of the chemical. Name CASRN The Chemical Abstract Service Registry Number for this chemical. TEEL-0 This is the threshold concentration below which most people will experience no appreciable risk of health effects. This PAC is always based on TEEL-0 because AEGL-0 or ERPG-0 values do not exist. PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value. PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-2 value. PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value. Units The units for the PAC values (ppm or mg/m3). Comments and Technical comments by the developers of the PAC data set. -

List of Designated Additives
The Japan Food chemical Research Faundation List of Designated Additives The substances below are the designated additives appearing in Table 1, as mentioned in Article 12 of the Enforcement Regulations under the Food Sanitation Law. These additives are listed here in alphabetic order. They are 455 in total as of July 03, 2018. The number preceding the name of each additive is the sequence number given to the corresponding additive in the original Japanese table. 16 Acesulfame Potassium(Acesulfame K) 20 Acetaldehyde 322 Acetic Acid, Glacial 23 Acetone 22 Acetophenone 17 Acetylated Distarch Adipate 19 Acetylated Distarch Phosphate 18 Acetylated Oxidized Starch 5 Adipic Acid 26 Advantame 32 DL-Alanine 190 Aliphatic Higher Alcohols 191 Aliphatic Higher Aldehydes (except those generally recognized as highly toxic) 192 Aliphatic Higher Hydrocarbons (except those generally recognized as highly toxic) 178 Allyl Cyclohexylpropionate 364 Allyl Hexanoate (Allyl Caproate) 53 Allyl Isothiocyanate (Volatile Oil of Mustard) 429 Aluminum Ammonium Sulfate (Crystal: Ammonium Alum, Desiccated: Burnt Ammonium Alum) 430 Aluminum Potassium Sulfate (Crystal: Alum or Potassium Alum, Desiccated: Burnt Alum) 29 (3-Amino-3-carboxypropyl)dimethylsulfonium chloride 43 Ammonia 35 Ammonium Alginate 240 Ammonium Bicarbonate (Ammonium Hydrogen Carbonate) 237 Ammonium Carbonate 81 Ammonium Chloride 446 Ammonium Dihydrogen Phosphate (Ammonium Phosphate, Monobasic or Monoammonium Phosphate) 44 Ammonium isovalerate 98 Ammonium Persulfate 431 Ammonium Sulfate 30 Amylalcohol -

Sodium Hexametaphosphate from China
Sodium Hexametaphosphate From China Investigation No. 731-TA-1110 (Final) Publication 3984 March 2008 Washington, DC 20436 U.S. International Trade Commission COMMISSIONERS Daniel R. Pearson, Chairman Shara L. Aranoff, Vice Chairman Deanna Tanner Okun Charlotte R. Lane Irving A. Williamson Dean A. Pinkert Robert A. Rogowsky Director of Operations Staff assigned Debra Baker, Investigator Philip Stone, Industry Analyst Craig Thomsen, Economist John Ascienzo, Auditor Robin Turner, Attorney Mara Alexander, Statistician George Deyman, Supervisory Investigator Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Sodium Hexametaphosphate From China Investigation No. 731-TA-1110 (Final) Publication 3984 March 2008 CONTENTS Page Determination................................................................... 1 Views of the Commission ......................................................... 3 Part I: Introduction .............................................................. I-1 Background .................................................................. I-1 Organization of the report....................................................... I-1 Summary data................................................................ I-2 Previous and related investigations................................................ I-3 Nature and extent of sales at LTFV ............................................... I-3 The subject -

Safety Assessment of Phosphoric Acid and Simple Salts As Used in Cosmetics
Safety Assessment of Phosphoric Acid and Simple Salts as Used in Cosmetics Status: Scientific Literature Review for Public Comment Release Date: November, 2015 Panel Date: March 31-April 1, 2016 All interested persons are provided 60 days from the above date to comment on this safety assessment and to identify additional published data that should be included or provide unpublished data which can be made public and included. Information may be submitted without identifying the source or the trade name of the cosmetic product containing the ingredient. All unpublished data submitted to CIR will be discussed in open meetings, will be available at the CIR office for review by any interested party and may be cited in a peer-reviewed scientific journal. Please submit data, comments, or requests to the CIR Director, Dr. Lillian J. Gill. The 2015 Cosmetic Ingredient Review Expert Panel members are: Chair, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Ronald A. Hill, Ph.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; James G. Marks, Jr., M.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Director is Lillian J. Gill, D.P.A. This report was prepared by Wilbur Johnson, Jr., M.S., Senior Scientific Analyst and Bart Heldreth, Ph.D., Chemist. © Cosmetic Ingredient Review 1620 L STREET, NW, SUITE 1200 ◊ WASHINGTON, DC 20036-4702 ◊ PH 202.331.0651 ◊ FAX 202.331.0088 ◊ [email protected] INTRODUCTION The safety of the following 31 ingredients, phosphoric acid and its salts, as used -

Commission Directive 2002/82/EC Amending Directive 96
28.10.2002 EN Official Journal of the European Communities L 292/1 I (Acts whose publication is obligatory) COMMISSION DIRECTIVE 2002/82/EC of 15 October 2002 amending Directive 96/77/EC laying down specific purity criteria on food additives other than colours and sweeteners (Text with EEA relevance) THE COMMISSION OF THE EUROPEAN COMMUNITIES, (4) It is necessary to take into account the specifications and analytical techniques for additives as set out in the Codex Alimentarius as drafted by the Joint FAO/WHO Expert Having regard to the Treaty establishing the European Committee on Food Additives (JECFA). Community, (5) Directive 96/77/EC should therefore be amended accordingly. Having regard to Council Directive 89/107/EEC of 21 December 1988 on the approximation of the laws of the Member States concerning food additives authorised for use in (6) The measures provided for in this Directive are in foodstuffs intended for human consumption (1), as amended accordance with the opinion of the Standing Committee by Directive 94/34/EC of the European Parliament and of the on the Food Chain and Animal Health, Council (2) and in particular Article 3(3)(a) thereof, HAS ADOPTED THIS DIRECTIVE: After consulting the Scientific Committee on Food, Whereas: Article 1 The Annex to Directive 96/77/EC is amended as set out in the (1) Directive 95/2/EC of the European Parliament and of Annex to this Directive. the Council of 20 February 1995 on food additives other than colours and sweeteners (3), as last amended by Directive 2001/5/EC (4), lists those substances which may be used as additives other than colours and Article 2 sweeteners in foodstuffs. -
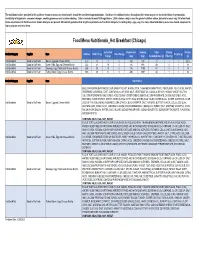
Food Menu Nutritional Summary by Region 2.20.2020.Xlsx
The nutritional values provided in this guide or shown on menus or menu boards should be considered approximations. Variations in nutritional values throughout this menu may occur due to deviations in preparation, availability of ingredients, seasonal changes, cooking processes and custom ordering. Data is rounded to meet FDA regulations. 2,000 calories a day is used for general nutrition advice, but calorie needs vary. All of our food items are produced in facilities where known allergens are present. We cannot guarantee that any of our products are free from allergens (including dairy, eggs, soy, tree nuts, wheat and others) as we use shared equipment to store, prepare and serve them. Food Menu Nutritionals_Hot Breakfast (Chicago) Saturated Cholesterol Sodium Total Dietary Protein Product Category Supplier Item Calories Total Fat (g) Trans Fat (g) Sugars (g) Fat (g) (mg) (mg) Carbohydrates (g) Fiber (g) (g) Hot Breakfast Good to Go Fresh Bacon, Egg and Cheese Muffin 420 18 7 0 160 730 31 1 1 20.5 Hot Breakfast Good to Go Fresh Green Chile, Egg and Cheese Burrito 522 21 10 0 136 993 49 3 1 18 Hot Breakfast Good to Go Fresh Sausage, Egg, Potato and Cheese Burrito 505 30 13 0 116 1190 41 2 1 23 Hot Breakfast Good to Go Fresh Turkey Bacon, Egg, Cheese Burrito 600 29 11 0 200 1567 44 1 1 36 Product Category Supplier Item Ingredients ENGLISH MUFFIN (ENRICHED FLOUR (WHEAT FLOUR, NIACIN, IRON, THIAMINE MONONITRATE, RIBOFLAVIN, FOLIC ACID), WATER, DEGERMED CORNMEAL, EAST, CONTAINS 2% OR LESS: SALT, VEGETABLE OIL (CANOLA OR SOY), SUGAR, WHEAT -

Sodium Hexametaphosphate from China
Sodium Hexametaphosphate From China Investigation No. 731-TA-1110 (Preliminary) Publication 3912 April 2007 U.S. International Trade Commission COMMISSIONERS Daniel R. Pearson, Chairman Shara L. Aranoff, Vice Chairman Deanna Tanner Okun Charlotte R. Lane Irving A. Williamson Dean A. Pinkert Robert A. Rogowsky Director of Operations Staff assigned Debra Baker, Investigator Philip Stone, Industry Analyst Craig Thomsen, Economist John Ascienzo, Accountant/Auditor Robin Turner, Attorney George L. Deyman, Supervisory Investigator Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Sodium Hexametaphosphate From China Investigation No. 731-TA-1110 (Preliminary) Publication 3912 April 2007 CONTENTS Page Determination................................................................... 1 Views of the Commission ......................................................... 3 Separate and additional views of Chairman Daniel R. Pearson and Commissioner Deanna Tanner Okun concerning Bratsk Aluminum v. United States .................................. 19 Part I: Introduction .............................................................. I-1 Background .................................................................. I-1 Organization of the report....................................................... I-1 Summary data................................................................ I-2 Previous and related investigations............................................... -

Calcium Chloride in the "Calcium Chloride Process" (21 CFR 184.1191)
Phosphates Handling/Processing 1 2 Identification of Petitioned Substances 3 This report addresses the following phosphate salts allowed under the National Organic Program (NOP) 4 regulations at 7 CFR 205.605(b): calcium phosphates (monobasic, dibasic and tribasic), potassium 5 phosphate, sodium acid pyrophosphate, and sodium phosphates. Chemical identifications of these 6 phosphates are included in Table 1. 7 8 Table 1: Chemical Identification of the Phosphates Listed at 7 CFR 205.605(b). Chemical Names Chemical Formula CAS Nos. E/INS No. Calcium phosphate, monobasic Calcium dihydrogen phosphate Ca(H2PO4)2 (anhydrous) 7758-23-8 Calcium biphosphate Calcium bis(dihydrogen phosphate) E 341(i) Monocalcium phosphate Primary calcium phosphate Ca(H2PO4)2 · 1 H2O 10031-30-8 Acid calcium phosphate Calcium diorthophosphate Calcium phosphate, dibasic CaHPO4 (anhydrous) 7757-93-9 Calcium hydrogen phosphate E 341(ii) Monocalcium acid phosphate CaHPO4· 2 H2O 7789-77-7 Dicalcium orthophosphate Calcium phosphate, tribasic Tricalcium diphosphate Ca3(PO4)2 (anhydrous) 7758-87-4 E 341(iii) Tricalcium phosphate Tricalcium orthophosphate Dipotassium phosphate (anhydrous) Dipotassium hydrogen phosphate K2HPO4 (anhydrous) 7758-11-4 Potassium hydrogen phosphate E 340(ii) Potassium dibasic phosphate K2HPO4· 3 H2O 16788-57-1 Potassium phosphate dibasic Sodium acid pyrophosphate (SAPP) Disodium diphosphate Na2H2P2O7 (anhydrous) 7758-16-9 E 450(vi) Disodium dihydrogen pyrophosphate; Diphosphoric acid, disodium salt Monosodium phosphate 7558-80-7 NaH2PO4 (anhydrous) -

Safety Assessment of Phosphoric Acid and Its Salts As Used in Cosmetics
Safety Assessment of Phosphoric Acid and Its Salts as Used in Cosmetics Status: Final Report Release Date: November 16, 2016 Panel Date: September 26-27, 2016 The 2016 Cosmetic Ingredient Review Expert Panel members are: Chair, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Ronald A. Hill, Ph.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; James G. Marks, Jr., M.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Director is Lillian J. Gill, D.P.A. This report was prepared by Wilbur Johnson, Jr., M.S., Senior Scientific Analyst, Ivan Boyer, Ph.D., Toxicologist, and Bart Heldreth, Ph.D., Chemist. © Cosmetic Ingredient Review 1620 L STREET, NW, SUITE 1200 ◊ WASHINGTON, DC 20036-4702 ◊ PH 202.331.0651 ◊ FAX 202.331.0088 ◊ [email protected] ABSTRACT: The Cosmetic Ingredient Review (CIR) Expert Panel (Panel) reviewed the safety of Phosphoric Acid and its salts (31 ingredients), which function as buffering agents, corrosion inhibitors, chelating agents, and pH adjusters in cosmetic products. The Panel reviewed data relating to the safety of these ingredients, and concluded that Phosphoric Acid and its salts are safe in the present practices of use and concentration in cosmetics when formulated to be non-irritating. INTRODUCTION The safety of the following 31 ingredients, as used in cosmetics (with systematic nomenclature in parenthesis when different from the ingredient name), is reviewed in this safety assessment: Phosphoric Acid • (calcium hydrogen Phosphate Buffered Saline -

SODIUM ACID PYROPHOSPHATE (SAPP) in Produce
PETITION TO THE NATIONAL ORGANIC STANDARDS BOARD For the Use of SODIUM ACID PYROPHOSPHATE (SAPP) In Produce Item A: This Petition supports the International Food Additives Council’s (IFAC) request to amend the National List for “Nonagricultural (nonorganic) substances allowed in or on processed products labeled as ‘organic’ or made with organic (specified ingredients)” under Section 205.605(b). This Petition relates to Sodium Acid Pyrophosphate (SAPP) as described in the Food Chemicals Codex (FCC), as amended, including alternative nomenclature included therein, as described below. Currently, the National List under Section 205.605(b) provides for the use of SAPP, “for use only as a leavening agent.” This Petition requests an additional use for this substance, namely as a sequestrant to maintain the appearance of cooked and uncooked fruits and vegetables. Item B: (1) COMMON NAME Sodium Acid Pyrophosphate: (OTHER NAMES: Disodium Pyrophosphate; Disodium Dihydrogen Pyrophosphate; Acid Sodium Pyrophosphate; Disodium Diphosphate). (2) CONTACT INFORMATION This Petition is submitted by the International Food Additives Council (IFAC), an international association representing manufacturers and suppliers of high quality substances used worldwide as food ingredients. In particular, IFAC’s Phosphates Committee is comprised of domestic and global suppliers of phosphates, used worldwide in food products. The information contained in this Petition was compiled using information provided by the IFAC Phosphates Committee members. Contact Information: Lyn O’Brien Nabors President International Food Additives Council 1100 Johnson Ferry Road Suite 300 Atlanta, GA 30342 Phone: 404-252-3663 Fax: 404-252-0774 Email: [email protected] (3) INTENDED USE OF THE SUBSTANCE As stated above, the purpose of this Petition is to add another use, in addition to leavening in baked goods, for this substance, namely as a sequestrant on cooked or uncooked produce to maintain the appearance and texture of these foods.