Comparison of Glimepiride, Alogliptin and Alogliptin+Pioglitazone Combination in Poorly Controlled Type 2 Diabetic Patients ( Protocol: Takeda ALO-IIT)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Optum Essential Health Benefits Enhanced Formulary PDL January
PENICILLINS ketorolac tromethamineQL GENERIC mefenamic acid amoxicillin/clavulanate potassium nabumetone amoxicillin/clavulanate potassium ER naproxen January 2016 ampicillin naproxen sodium ampicillin sodium naproxen sodium CR ESSENTIAL HEALTH BENEFITS ampicillin-sulbactam naproxen sodium ER ENHANCED PREFERRED DRUG LIST nafcillin sodium naproxen DR The Optum Preferred Drug List is a guide identifying oxacillin sodium oxaprozin preferred brand-name medicines within select penicillin G potassium piroxicam therapeutic categories. The Preferred Drug List may piperacillin sodium/ tazobactam sulindac not include all drugs covered by your prescription sodium tolmetin sodium drug benefit. Generic medicines are available within many of the therapeutic categories listed, in addition piperacillin sodium/tazobactam Fenoprofen Calcium sodium to categories not listed, and should be considered Meclofenamate Sodium piperacillin/tazobactam as the first line of prescribing. Tolmetin Sodium Amoxicillin/Clavulanate Potassium LOW COST GENERIC PREFERRED For benefit coverage or restrictions please check indomethacin your benefit plan document(s). This listing is revised Augmentin meloxicam periodically as new drugs and new prescribing LOW COST GENERIC naproxen kit information becomes available. It is recommended amoxicillin that you bring this list of medications when you or a dicloxacillin sodium CARDIOVASCULAR covered family member sees a physician or other penicillin v potassium ACE-INHIBITORS healthcare provider. GENERIC QUINOLONES captopril ANTI-INFECTIVES -

The Genetics of Adverse Drug Outcomes in Type 2 Diabetes: a Systematic Review
SYSTEMATIC REVIEW published: 14 June 2021 doi: 10.3389/fgene.2021.675053 The Genetics of Adverse Drug Outcomes in Type 2 Diabetes: A Systematic Review Assefa M. Baye 1, Teferi G. Fanta 1, Moneeza K. Siddiqui 2 and Adem Y. Dawed 2* 1 Department of Pharmacology and Clinical Pharmacy, College of Health Sciences, Addis Ababa University, Addis Ababa, Ethiopia, 2 Division of Population Health and Genomics, Ninewells Hospital and School of Medicine, University of Dundee, Dundee, United Kingdom Background: Adverse drug reactions (ADR) are a major clinical problem accounting for significant hospital admission rates, morbidity, mortality, and health care costs. One-third of people with diabetes experience at least one ADR. However, there is notable interindividual heterogeneity resulting in patient harm and unnecessary medical costs. Genomics is at the forefront of research to understand interindividual variability, and there are many genotype-drug response associations in diabetes with inconsistent findings. Here, we conducted a systematic review to comprehensively examine and synthesize the effect of genetic polymorphisms on the incidence of ADRs of oral glucose-lowering drugs in people with type 2 diabetes. Edited by: Celine Verstuyft, Methods: A literature search was made to identify articles that included specific Université Paris-Saclay, France results of research on genetic polymorphism and adverse effects associated with Reviewed by: oral glucose-lowering drugs. The electronic search was carried out on 3rd October Zhiguo Xie, 2020, through Cochrane Library, PubMed, and Web of Science using keywords and Central South University, China Vera Ribeiro, MeSH terms. University of Algarve, Portugal Result: Eighteen articles consisting of 10, 383 subjects were included in this review. -
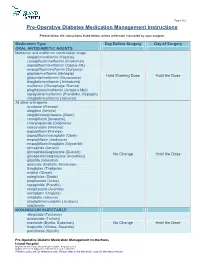
Pre-Operative Diabetes Medication Management Instructions
Page 1 of 2 Pre-Operative Diabetes Medication Management Instructions Please follow the instructions listed below unless otherwise instructed by your surgeon Medication Type Day Before Surgery Day of Surgery ORAL ANTIDIABETIC AGENTS Metformin and metformin combination drugs alogliptin/metformin (Kazano) canagliflozin/metformin (Invokamet) dapagliflozin/metformin (Xigduo XR) empagliflozin/metformin (Synjardy) glipizide/metformin (Metaglip) Hold Evening Dose Hold the Dose glyburide/metformin (Glucovance) linagliptin/metformin (Jentadueto) metformin (Glucophage, Riomet) pioglitazone/metformin (Actoplus Met) repaglidine/metformin (PrandiMet, Repaglin) sitagliptin/metformin (Janumet) All other oral agents acarbose (Precose) alogliptin (Nesina) alogliptin/pioglitazone (Oseni) canagliflozin (Invokana) chlorpropamide (Diabinese) colesevelam (Welchol) dapagliflozin (Farxiga) dapagliflozin/saxagliptin (Qtern) empagliflozin (Jardiance) empagliflozin/linagliptin (Glyxambi) glimepiride (Amaryl) glimepiride/pioglitazone (Duetact) No Change Hold the Dose glimepiride/rosiglitazone (Avandaryl) glipizide (Glucotrol) glyburide (DiaBeta, Micronase) linagliptan (Tradjenta) miglitol (Glyset) nateglinide (Starlix) pioglitazone (Actos) repaglinide (Prandin) rosiglitazone (Avandia) saxagliptin (Onglyza) sitagliptin (Januvia) sitagliptin/simvastatin (Juvisync) tolazamide NON-INSULIN INJECTABLE albiglutide (Tanzeum) dulaglutide (Trulicity) exenatide (Byetta, Bydureon) No Change Hold the Dose liraglutide (Victoza, Saxenda) pramlintide (Symlin) Pre-Operative Diabetic -

OSENI (Alogliptin and Pioglitazone) Tablets II May Increase Risk
HIGHLIGHTS OF PRESCRIBING INFORMATION -----------------------WARNINGS AND PRECAUTIONS--------------------- These highlights do not include all the information needed to use • Congestive heart failure: Fluid retention may occur and can OSENI safely and effectively. See full prescribing information for exacerbate or lead to congestive heart failure. Combination use OSENI. with insulin and use in congestive heart failure NYHA Class I and OSENI (alogliptin and pioglitazone) tablets II may increase risk. Monitor patients for signs and symptoms. Initial U.S. Approval: 2013 (5.1) • Acute pancreatitis: There have been postmarketing reports of WARNING: CONGESTIVE HEART FAILURE acute pancreatitis. If pancreatitis is suspected, promptly See full prescribing information for complete boxed warning discontinue OSENI. (5.2) • Thiazolidinediones, including pioglitazone, cause or • Hypersensitivity: There have been postmarketing reports of exacerbate congestive heart failure in some patients. (5.1) serious hypersensitivity reactions in patients treated with alogliptin • After initiation of OSENI and after dose increases, monitor such as anaphylaxis, angioedema and severe cutaneous adverse patients carefully for signs and symptoms of heart failure reactions. In such cases, promptly discontinue OSENI, assess for (e.g., excessive, rapid weight gain, dyspnea and/or other potential causes, institute appropriate monitoring and edema). If heart failure develops, it should be managed treatment and initiate alternative treatment for diabetes. (5.3) according to current standards of care and • Hepatic effects: Postmarketing reports of hepatic failure, discontinuation or dose reduction of pioglitazone in OSENI sometimes fatal. Causality cannot be excluded. If liver injury is must be considered. detected, promptly interrupt OSENI and assess patient for • OSENI is not recommended in patients with symptomatic probable cause, then treat cause if possible, to resolution or heart failure. -

Sulfonylureas
Therapeutic Class Overview Sulfonylureas INTRODUCTION In the United States (US), diabetes mellitus affects more than 30 million people and is the 7th leading cause of death (Centers for Disease Control and Prevention [CDC] 2018). Type 2 diabetes mellitus (T2DM) is the most common form of diabetes and is characterized by elevated fasting and postprandial glucose concentrations (American Diabetes Association [ADA] 2019[a]). It is a chronic illness that requires continuing medical care and ongoing patient self-management education and support to prevent acute complications and to reduce the risk of long-term complications (ADA 2019[b]). ○ Complications of T2DM include hypertension, heart disease, stroke, vision loss, nephropathy, and neuropathy (ADA 2019[a]). In addition to dietary and lifestyle management, T2DM can be treated with insulin, one or more oral medications, or a combination of both. Many patients with T2DM will require combination therapy (Garber et al 2019). Classes of oral medications for the management of blood glucose levels in patients with T2DM focus on increasing insulin secretion, increasing insulin responsiveness, or both, decreasing the rate of carbohydrate absorption, decreasing the rate of hepatic glucose production, decreasing the rate of glucagon secretion, and blocking glucose reabsorption by the kidney (Garber et al 2019). Pharmacologic options for T2DM include sulfonylureas (SFUs), biguanides, thiazolidinediones (TZDs), meglitinides, alpha-glucosidase inhibitors, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 (GLP-1) analogs, amylinomimetics, sodium-glucose cotransporter 2 (SGLT2) inhibitors, combination products, and insulin (Garber et al 2019). SFUs are the oldest of the oral antidiabetic medications, and all agents are available generically. The SFUs can be divided into 2 categories: first-generation and second-generation. -

NESINA (Alogliptin) Tablets, for Oral Use • Heart Failure: Consider the Risks and Benefits of NESINA Prior to Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION ------------------------WARNINGS AND PRECAUTIONS---------------------- These highlights do not include all the information needed to use • Acute pancreatitis: There have been postmarketing reports of NESINA safely and effectively. See full prescribing information for acute pancreatitis. If pancreatitis is suspected, promptly NESINA. discontinue NESINA. (5.1) NESINA (alogliptin) tablets, for oral use • Heart failure: Consider the risks and benefits of NESINA prior to Initial U.S. Approval: 2013 initiating treatment in patients at risk for heart failure. If heart failure develops, evaluate and manage according to current ---------------------------RECENT MAJOR CHANGES-------------------------- standards of care and consider discontinuation of NESINA (5.2). Indications and Usage (1.1) 4/2016 • Hypersensitivity: There have been postmarketing reports of Dosage and Administration serious hypersensitivity reactions in patients treated with NESINA Patients with Renal Impairment (2.2) 4/2016 such as anaphylaxis, angioedema and severe cutaneous adverse Warnings and Precautions reactions, including Stevens-Johnson syndrome. In such cases, Pancreatitis (5.1) 4/2016 promptly discontinue NESINA, assess for other potential causes, Heart Failure (5.2) 4/2016 institute appropriate monitoring and treatment and initiate Hepatic Effects (5.4) 4/2016 alternative treatment for diabetes. (5.3) Bullous Pemphigoid (5.7) 12/2016 • Hepatic effects: Postmarketing reports of hepatic failure, ----------------------------INDICATIONS AND USAGE--------------------------- sometimes fatal. Causality cannot be excluded. If liver injury is NESINA is a dipeptidyl peptidase-4 (DPP-4) inhibitor indicated as an detected, promptly interrupt NESINA and assess patient for adjunct to diet and exercise to improve glycemic control in adults with probable cause, then treat cause if possible, to resolution or type 2 diabetes mellitus. -

Diabetes Recommendations and Tier Coverage Chart
DIABETES RECOMMENDATIONS AND TIER COVERAGE CHART The American Diabetes Association guidelines for 2020, recommend metformin as the preferred initial treatment for type 2 diabetes (T2DM) along with weight management and physical activity. In patients who have established ASVD or at high risk, CKD, or HF, a SGLT2i or GLP-1 receptor with proven efficacy is recommended independent of A1C. • ASCVD dominates: o GLP-1RA with proven CVD benefit (dulaglutide, liraglutide, injectable semaglutide) OR o SGLT2i with proven CVD benefit (canagliflozin, empagliflozin) if adequate eGFR • HF or CKD dominates: o SGLT2i with evidence of reducing HF and/or CKD progression (empagliflozin, canagliflozin, dapagliflozin) if adequate eGFR OR o If SGLT2i intolerant/contraindicated or eGFR is inadequate, then GLP-1RA with proven CVD benefit In individuals without established cardiovascular disease, pharmacological treatment should be patient-centered taking into account side-effects, cost, impact on weight, risk of hypoglycemia, and other patient preferences. For more detailed information regarding ADA recommendations for pharmacological agents to treat T2DM click here. The following chart is a list of oral and injectable diabetes medications listed by class with their respective A1C reduction and insurance coverage and/or coverage requirements for BCBS, HPHC, Tufts, TMP, and MassHealth. Tufts Medicare Medications BCBSMA HPHC Tufts Preferred MassHealth Biguanides A1C reduction: 1-1.5% metformin Tier 1 Tier 1;2 Tier 1 Tier 1 Covered Glucoghage (metformin) NC NC NC;Tier -

Initial Combination Therapy with Canagliflozin Plus Metformin Versus Each Component As Monotherapy for Drug-Naïve Type 2 Diabe
Diabetes Care Volume 39, March 2016 353 Julio Rosenstock,1 Leonard Chuck,2 Initial Combination Therapy With Manuel Gonzalez-Ortiz,´ 3 Kate Merton,4 CLIN CARE/EDUCATION/NUTRITION/PSYCHOSOCIAL Jagriti Craig,4 George Capuano,4 and Canagliflozin Plus Metformin Rong Qiu4 Versus Each Component as Monotherapy for Drug-Na¨ıve Type 2 Diabetes Diabetes Care 2016;39:353–362 | DOI: 10.2337/dc15-1736 OBJECTIVE This study assessed the efficacy/safety of canagliflozin (CANA), a sodium–glucose cotransporter 2 (SGLT2) inhibitor, plus metformin extended-release (MET) initial therapy in drug-na¨ıve type 2 diabetes. RESEARCH DESIGN AND METHODS This 26-week, double-blind, phase 3 study randomized 1,186 patients to CANA 100 mg (CANA100)/MET, CANA 300 mg (CANA300)/MET, CANA100, CANA300, or MET. Primary end point was change in HbA1c at week 26 for combinations versus monotherapies. Secondary end points included noninferiority in HbA1c lowering with CANA monotherapy versus MET; changes in fasting plasma glucose, body weight, and blood pressure; and proportion of patients achieving HbA1c <7.0% (<53 mmol/mol). RESULTS From mean baseline HbA1c of 8.8% (73 mmol/mol), CANA100/MET and CANA300/ MET significantly lowered HbA1c versus MET (median dose, 2,000 mg/day) by –1.77%, –1.78%, and –1.30% (–19.3, –19.5, and –14.2 mmol/mol; differences of 1Dallas Diabetes and Endocrine Center at Medi- 2 – – – P cal City, Dallas, TX 0.46% and 0.48% [ 5.0 and 5.2 mmol/mol]; = 0.001) and versus CANA100 2 – – – – Diablo Clinical Research, Walnut Creek, CA and CANA300 by 1.37% and 1.42% ( 15.0 and 15.5 mmol/mol; differences of 3Institute of Experimental and Clinical Therapeu- –0.40% and –0.36% [–4.4 and –3.9 mmol/mol]; P = 0.001). -

Short-Term Efficacy and Safety of Repaglinide Versus Glimepiride As
Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy Dovepress open access to scientific and medical research Open Access Full Text Article ORIGINAL RESEARCH Short-term efficacy and safety of repaglinide versus glimepiride as augmentation of metformin in treating patients with type 2 diabetes mellitus This article was published in the following Dove Press journal: Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy Jing Xie1 Background: Consistent evidence is still lacking on which one, glimepiride plus metformin Ning Li1 or repaglinide plus metformin, is better in treating type 2 diabetes mellitus (T2DM). Xiaoyan Jiang1 Therefore, this study was conducted to compare the short-term efficacy and safety of these Liyin Chai1 two methods in treating T2DM. Jian-Jun Chen2 Methods: The literature research dating up to August 2018 was conducted in the electronic Wuquan Deng1 databases. The randomized controlled trials (RCTs) comparing the short-term (treatment period ≤12 weeks) efficacy and safety of these two methods in treating patients with 1 Department of Endocrinology and T2DM were included. No language limitation was used in this study. The decreased Nephrology, Chongqing University Central Hospital, Chongqing Emergency hemoglobin A1c (HbA1c), fasting plasma glucose (FPG), and 2h plasma glucose (2hPG) Medical Center, Chongqing 400014, levels were used as the primary outcome to assess the efficacy, and the adverse events and ’ 2 People s Republic of China; Institute of hypoglycemia were used as the secondary outcome to assess the safety. Life Sciences, Chongqing Medical University, Chongqing 400016, People’s Results: In total, 11 RCTs composed of 844 T2DM patients were included. The results Republic of China showed that there were no significant differences in decreasing HbA1c and FPG levels between the two methods, but the estimated standardized mean differences favored the repaglinide plus metformin. -

Combination Use of Insulin and Incretins in Type 2 Diabetes
Canadian Agency for Agence canadienne Drugs and Technologies des médicaments et des in Health technologies de la santé CADTH Optimal Use Report Volume 3, Issue 1C Combination Use of Insulin and July 2013 Incretins in Type 2 Diabetes Supporting Informed Decisions This report is prepared by the Canadian Agency for Drugs and Technologies in Health (CADTH). The report contains a comprehensive review of the existing public literature, studies, materials, and other information and documentation (collectively the “source documentation”) available to CADTH at the time of report preparation. The information in this report is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. The information in this report should not be used as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process, nor is it intended to replace professional medical advice. While CADTH has taken care in the preparation of this document to ensure that its contents are accurate, complete, and up to date as of the date of publication, CADTH does not make any guarantee to that effect. CADTH is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in the source documentation. CADTH is not responsible for any errors or omissions or injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the information in this document or in any of the source documentation. -

ANTI-HYPERGLYCEMIC DIABETES AGENTS in T2DM: Color Outcomes Comparison Summary Table
ANTI-HYPERGLYCEMIC DIABETES AGENTS in T2DM: Outcomes Comparison Summary Table L Regier BSP BA, M LeBras PharmD, T Trischuk PharmD, J Bareham BSP, L Lu BSP © www.RxFiles.ca Aug 2021 Drug Class Sulfonylureas TZDs Meglitinides DPP4 Inhibitors GLP1 Agonists *** SGLT2 Inhibitors *** Insulin in T2DM Generic Metformin Gliclazide Glyburide Pioglitazone Rosiglitazone Acarbose Repaglinide Saxagliptin ONGLYZA Liraglutide VICTOZA Empagliflozin JARDIANCE Intensity: Intensity: BRAND (MF) DIAMICRON DIABETA ACTOS, g AVANDIA GLUCOBAY GLUCONORM Sitagliptin JANUVIA Exenatide BYETTA, BYDUREON Canagliflozin INVOKANA Less More GLUCOPHAGE Dulaglutide TRULICITY Dapagliflozin FORXIGA, FARXIGA Alogliptin NESINA (e.g. NPH (Multiple daily GLUCOTROL D/C STEGLATRO Glipizide Nateglinide Semaglutide OZEMPIC, RYBELSUS (PO) Ertugliflozin Linagliptin TRAJENTA HS + MF) doses) STARLIX D/C SPREAD-DIMCAD] Lixisenatide ADLYXINE; ALBIGLUTIDE D/C SAVOR-TIMI 53, EMPA-REG, CANVAS, CREDENCE, T2DM: UKPDS-33,80; ADVANCE, Major trials to UKPDS- ProACTIVE ACE 33,34,80 UKPDS- Meta-analysis. TECOS, EXAMINE LEADER, EXSCEL, FREEDOM CVO, DECLARE, VERTIS-CV (2020), ACCORD, VADT, ORIGIN, DEVOTE support ADVANCE (Prevention (ADOPT; 33,80 Ferwana M. Meta- RECORD interim, PROLOGUE, REWIND, SUSTAIN-6, PIONEER-6, DAPA-HF, DAPA-CKD (2020), T1DM: DCCT/EDIC findings/ analysis 2013. trial: Stop- - some use in (ADOPT) ADOPT, DREAM CARMELINA, EMPEROR-Reduced & -Preserved (Also Boussageon et al. Meta- Outcomes* ADVANCE) SR-Liao 2017; IRIS NIDDM) ELIXA, HARMONY CAROLINA (2020), EMPA-Kidney (2022) analysis. -

AVANDARYL™ (Rosiglitazone Maleate and Glimepiride) Tablets
AA:LX PRESCRIBING INFORMATION AVANDARYL™ (rosiglitazone maleate and glimepiride) Tablets DESCRIPTION AVANDARYL (rosiglitazone maleate and glimepiride) tablets contain 2 oral antidiabetic drugs used in the management of type 2 diabetes: Rosiglitazone maleate and glimepiride. Rosiglitazone maleate is an oral antidiabetic agent of the thiazolidinedione class which acts primarily by increasing insulin sensitivity. Rosiglitazone maleate is not chemically or functionally related to the sulfonylureas, the biguanides, or the alpha-glucosidase inhibitors. Chemically, rosiglitazone maleate is (±)-5-[[4-[2-(methyl-2-pyridinylamino) ethoxy]phenyl] methyl]-2,4-thiazolidinedione, (Z)-2-butenedioate (1:1) with a molecular weight of 473.52 (357.44 free base). The molecule has a single chiral center and is present as a racemate. Due to rapid interconversion, the enantiomers are functionally indistinguishable. The molecular formula is C18H19N3O3S•C4H4O4. Rosiglitazone maleate is a white to off-white solid with a melting point range of 122° to 123°C. The pKa values of rosiglitazone maleate are 6.8 and 6.1. It is readily soluble in ethanol and a buffered aqueous solution with pH of 2.3; solubility decreases with increasing pH in the physiological range. The structural formula of rosiglitazone maleate is: Glimepiride is an oral antidiabetic drug of the sulfonylurea class. Glimepiride is a white to yellowish-white, crystalline, odorless to practically odorless powder. Chemically, glimepiride is 1-[[p-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido)ethyl]phenyl]sulfonyl]-3-(trans-4- methylcyclohexyl)urea with a molecular weight of 490.62. The molecular formula for glimepiride is C24H34N4O5S. Glimepiride is practically insoluble in water.