Evolution, Expression and Meiotic Behavior of Genes Involved in Chromosome Segregation of Monotremes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evolution, Expression and Meiotic Behavior of Genes Involved in Chromosome Segregation of Monotremes
G C A T T A C G G C A T genes Article Evolution, Expression and Meiotic Behavior of Genes Involved in Chromosome Segregation of Monotremes Filip Pajpach , Linda Shearwin-Whyatt and Frank Grützner * School of Biological Sciences, The University of Adelaide, Adelaide, SA 5005, Australia; fi[email protected] (F.P.); [email protected] (L.S.-W.) * Correspondence: [email protected] Abstract: Chromosome segregation at mitosis and meiosis is a highly dynamic and tightly regulated process that involves a large number of components. Due to the fundamental nature of chromosome segregation, many genes involved in this process are evolutionarily highly conserved, but duplica- tions and functional diversification has occurred in various lineages. In order to better understand the evolution of genes involved in chromosome segregation in mammals, we analyzed some of the key components in the basal mammalian lineage of egg-laying mammals. The chromosome passenger complex is a multiprotein complex central to chromosome segregation during both mitosis and meio- sis. It consists of survivin, borealin, inner centromere protein, and Aurora kinase B or C. We confirm the absence of Aurora kinase C in marsupials and show its absence in both platypus and echidna, which supports the current model of the evolution of Aurora kinases. High expression of AURKBC, an ancestor of AURKB and AURKC present in monotremes, suggests that this gene is performing all necessary meiotic functions in monotremes. Other genes of the chromosome passenger complex complex are present and conserved in monotremes, suggesting that their function has been preserved Citation: Pajpach, F.; in mammals. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -
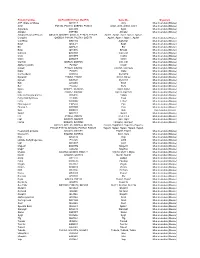
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Detection Rate. FC: Fold Change. GO: Gene Ontology. AUC: Area Under
Figure S1. Flow chart of the study. DR: detection rate. FC: fold change. GO: Gene Ontology. AUC: area under curve. KEGG: Kyoto Encyclopedia of Genes and Genomes. GEO: Gene Expression Omnibus. Figure S2. Independent validation of miRNAs and the classifier. A, ΔCt of 6 selected miRNAs in the independent cohort. *, P < 0.05. **, P < 0.01. ***, P < 0.001. B, Receiver operating curve of the classifier in the independent cohort. AUC: area under curve. Figure S3. Venn of predicted target genes from 3 platforms. Figure S4. GO enrichment analysis. GO: Gene Ontology. Table S1. miRNA expression profile in screening stage. Table S2. miRNA expression profile in validation stage. Table S3. Efficacy of the classifier. Table S4. miRNA expression profile in independent validation stage. Table S5. Predicted target genes. Table S6a. KEGG enrichment analysis. KEGG: Kyoto Encyclopedia of Genes and Genomes. Table S6b. GO_BP enrichment analysis. GO: Gene Ontology. BP: Biological Process. Table S6c. GO_CC enrichment analysis. CC: Cellular Component. Table S6d. GO_MF enrichment analysis. MF: Molecular Function. Table S7. GEO expression array datasets involved in this study. GEO: Gene Expression Omnibus. Table S8a. Predicted target genes covered by the selected GEO datasets. GEO: Gene Expression Omnibus. 1 Table S8b. Expression profiles of predicted target genes of hsa-miR-26b-5p in GEO datasets. Table S8c. Expression profiles of predicted target genes of hsa-miR-146b-5p in GEO datasets. Table S8d. Expression profiles of predicted target genes of hsa-miR-191-5p in GEO datasets. Table S8e. Expression profiles of predicted target genes of hsa-miR-484 in GEO datasets. -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures. -

Flotillin 1 Antibody (R31142)
Flotillin 1 Antibody (R31142) Catalog No. Formulation Size R31142 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability 1-3 business days Species Reactivity Human, Mouse, Rat Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Antigen affinity Buffer Lyophilized from 1X PBS with 2.5% BSA and 0.025% sodium azide/thimerosal UniProt O75955 Applications Western blot : 0.5-1ug/ml Limitations This Flotillin 1 antibody is available for research use only. Western blot testing of Flotillin 1 antbody; Lane 1: rat lung; 2: (r) brain; 3: (r) ovary; 4: human SMMC-7721; 5: (h) MFC-7 cell lysate. Expected/observed molecular weight ~49kDa. Description Flotillin 1 is a protein that in humans is encoded by the FLOT1 gene. The International Radiation Hybrid Mapping Consortium mapped the gene to chromosome 6. Bickel et al.(1997) found that mouse Flot1 behaves as a resident integral membrane protein of caveolae. It consistently copurified with Flot2 and with caveolin-1 in the purification of caveolin-rich membranes. Hazarika et al.(1999) found that stable transfection of Flot1, which they called ESA/flotillin-2, in COS-1 cells induced filopodia formation and changed the epithelial morphology to that of neuronal cells. Santamaria et al.(2005) found that prostate tumor overexpressed gene-1 interacted with Flotillin1 in detergent-insoluble membrane fractions. Flotillin1 colocalized with PTOV1 at the plasma membrane and in the nucleus, and it entered the nucleus concomitant with PTOV1 shortly before initiation of S phase. Application Notes The stated application concentrations are suggested starting amounts. -

Snipa Snpcard
SNiPAcard Block annotations Block info genomic range chr6:29,970,960-30,792,117 block size 821,158 bp variant count 107 variants Basic features Conservation/deleteriousness Linked genes μ = -0.141 [-3.802 – ABCF1 , AL662797.1 , AL662800.2 , ATAT1 , C6orf136 , DHX16 , FLOT1 4.141] , GNL1 , HCG17 , HCG18 , HCG19P , HCG20 , HCG4P3 , HLA-J , HLA-L , HLA-N , IER3 , LINC00243 , MDC1 , MICC , MRPS18B , NRM , PAIP1P1 , PPP1R10 , PPP1R11 , PPP1R18 , PTMAP1 , RN7SL353P , phyloP gene(s) hit or close-by RNF39 , RPP21 , SUCLA2P1 , TRIM10 , TRIM15 , TRIM26 , TRIM26BP , TRIM31 , TRIM31-AS1 , TRIM39 , TRIM39-RPP21 , TRIM40 , TUBB , UBQLN1P1 , XXbac-BPG252P9.10 , XXbac-BPG252P9.9 , XXbac-BPG283O16.9 , Y_RNA , ZNRD1 , ZNRD1-AS1 , ZNRD1-AS1_3 μ = 0.130 [0 – 1] BTN3A2 , BTN3A3 , C2 , CCHCR1 , CFB , FLOT1 , GABBR1 , HCG17 , HCG20 , HCG22 , HCP5 , HIST1H3I , HLA-A , HLA-B , HLA-C , HLA-F- AS1 , HLA-G , HLA-H , HLA-J , HLA-K , HLA-L , HLA-U , HLA-V , IER3 phastCons eQTL gene(s) , LINC00243 , MICB , NDUFS1 , OR5V1 , PPP1R18 , PSORS1C1 , RNF39 , RPP21 , TMEM154 , TRIM26BP , TRIM31 , TRIM39-RPP21 , VARS2 , WASF5P , ZFP57 , ZNRD1 , ZNRD1-AS1_3 μ = -0.462 [-9.98 – 5.1] ATAT1 , ATAT1 , ATAT1 , ATAT1 , ATAT1 , ATAT1 , ATAT1 , DDR1 , DDR1 , DDR1 , DDR1 , DDR1 , DDR1 , DDR1 , FLOT1 , FLOT1 , FLOT1 , FLOT1 , FLOT1 , FLOT1 , FLOT1 , FLOT1 , HCG19P , HCG19P potentially regulated , HCG19P , HCG19P , HCG19P , HCG19P , HCG19P , NRM , NRM , GERP++ gene(s) NRM , NRM , NRM , NRM , NRM , RNF39 , RNF39 , RNF39 , RNF39 , RNF39 , RNF39 , RNF39 , RNF39 , TRIM26 , TRIM26 , TRIM26 -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -

Evolutionarily Conserved Intercalated Disc Protein Tmem65 Regulates Cardiac Conduction and Connexin 43 Function
ARTICLE Received 11 Aug 2014 | Accepted 18 Aug 2015 | Published 25 Sep 2015 DOI: 10.1038/ncomms9391 Evolutionarily conserved intercalated disc protein Tmem65 regulates cardiac conduction and connexin 43 function Parveen Sharma1,*, Cynthia Abbasi1,*, Savo Lazic2, Allen C.T. Teng1, Dingyan Wang1, Nicole Dubois3, Vladimir Ignatchenko4, Victoria Wong5, Jun Liu6, Toshiyuki Araki4, Malte Tiburcy7, Cameron Ackerley8, Wolfram H. Zimmermann7, Robert Hamilton8,11, Yu Sun6, Peter P. Liu9, Gordon Keller3, Igor Stagljar5, Ian C. Scott2,8,11, Thomas Kislinger4,10 & Anthony O. Gramolini1,11 Membrane proteins are crucial to heart function and development. Here we combine cationic silica-bead coating with shotgun proteomics to enrich for and identify plasma membrane-associated proteins from primary mouse neonatal and human fetal ventricular cardiomyocytes. We identify Tmem65 as a cardiac-enriched, intercalated disc protein that increases during development in both mouse and human hearts. Functional analysis of Tmem65 both in vitro using lentiviral shRNA-mediated knockdown in mouse cardiomyocytes and in vivo using morpholino-based knockdown in zebrafish show marked alterations in gap junction function and cardiac morphology. Molecular analyses suggest that Tmem65 interaction with connexin 43 (Cx43) is required for correct localization of Cx43 to the intercalated disc, since Tmem65 deletion results in marked internalization of Cx43, a shorter half-life through increased degradation, and loss of Cx43 function. Our data demonstrate that the membrane protein Tmem65 is an intercalated disc protein that interacts with and functionally regulates ventricular Cx43. 1 Department of Physiology, University of Toronto, Toronto General Hospital Research Institute, Toronto, Ontario, Canada M5G 1L7. 2 Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada M5S 1A8. -

Fibroblasts from the Human Skin Dermo-Hypodermal Junction Are
cells Article Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling Valérie Haydont 1,*, Véronique Neiveyans 1, Philippe Perez 1, Élodie Busson 2, 2 1, 3,4,5,6, , Jean-Jacques Lataillade , Daniel Asselineau y and Nicolas O. Fortunel y * 1 Advanced Research, L’Oréal Research and Innovation, 93600 Aulnay-sous-Bois, France; [email protected] (V.N.); [email protected] (P.P.); [email protected] (D.A.) 2 Department of Medical and Surgical Assistance to the Armed Forces, French Forces Biomedical Research Institute (IRBA), 91223 CEDEX Brétigny sur Orge, France; [email protected] (É.B.); [email protected] (J.-J.L.) 3 Laboratoire de Génomique et Radiobiologie de la Kératinopoïèse, Institut de Biologie François Jacob, CEA/DRF/IRCM, 91000 Evry, France 4 INSERM U967, 92260 Fontenay-aux-Roses, France 5 Université Paris-Diderot, 75013 Paris 7, France 6 Université Paris-Saclay, 78140 Paris 11, France * Correspondence: [email protected] (V.H.); [email protected] (N.O.F.); Tel.: +33-1-48-68-96-00 (V.H.); +33-1-60-87-34-92 or +33-1-60-87-34-98 (N.O.F.) These authors contributed equally to the work. y Received: 15 December 2019; Accepted: 24 January 2020; Published: 5 February 2020 Abstract: Human skin dermis contains fibroblast subpopulations in which characterization is crucial due to their roles in extracellular matrix (ECM) biology.