The Age of Museomics How to Get Genomic Information from Museum Specimens of Lepidoptera Call, Elsa
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ulmaceae) at Huei-Sen Forests, Nantou Co
The LepidopterologicalSocietyLepidopterological Society of Japan ecLma bens. Iopid. Soc. IaPan 46 (4): 175-184, December 1995 The life histories and bielogy of Epicopeiidae of Taiwan Shen-Horn YEN", Jia-Horn Mu, Jia-Lurng JEAN Department of Entomology, National Chung-Hsing University, 250, Kuokuang Road., Taichung 402, Taiwan, R.O.C. Abstract The Iife histories and biology ef the twu species of Epicopeiidae of Taiwan are clescribed and illustratecl fer the first time. Additionally, a comment on phylogenetic position of this family is also provided. Key words Diurnal moths, immature stages, hostplants, phylogeny. Introduction The family Epicopeiidae embraces several medium- to large-sized diurnal moths across the Oriental and the Palaearctic regions. They are usually regarded as mimic to a certain species of Papilionidae, Danainae or bearing aposematic coloration. In Taiwan, two species of Epicopeiidae, EIPicopeia mencia Moore, 1874, and E. hainesii matsumuvai Okano, 1973, have hitherto been recognized. wnile the early stages of the same species from Japan and China have been described or illustrated, the life history of this family from Taiwan has remained unknown to date. In early May 1992, one of us, Yen, was able to collect larvae and eggs of E, mencia from Ulmus Parvijblia Jacq. (Ulmaceae) at Huei-Sen Forests, Nantou Co. and Shanping, Kaohsiung Co,, Taiwan, respectively. They were subsequently brought to the laboratory of Department of.Entomology. Rearing was conducted at room temperatures, Eggs were put into a small transparent plastic bag ; hatched larvae were reared in a 15 × 8 × 20 cm plastic case with an abundant supply of leaves of the hostplant; pupae were left in the case to emerge; hibernate pupae were mantained at 100C. -
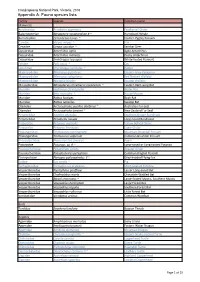
Report-VIC-Croajingolong National Park-Appendix A
Croajingolong National Park, Victoria, 2016 Appendix A: Fauna species lists Family Species Common name Mammals Acrobatidae Acrobates pygmaeus Feathertail Glider Balaenopteriae Megaptera novaeangliae # ~ Humpback Whale Burramyidae Cercartetus nanus ~ Eastern Pygmy Possum Canidae Vulpes vulpes ^ Fox Cervidae Cervus unicolor ^ Sambar Deer Dasyuridae Antechinus agilis Agile Antechinus Dasyuridae Antechinus mimetes Dusky Antechinus Dasyuridae Sminthopsis leucopus White-footed Dunnart Felidae Felis catus ^ Cat Leporidae Oryctolagus cuniculus ^ Rabbit Macropodidae Macropus giganteus Eastern Grey Kangaroo Macropodidae Macropus rufogriseus Red Necked Wallaby Macropodidae Wallabia bicolor Swamp Wallaby Miniopteridae Miniopterus schreibersii oceanensis ~ Eastern Bent-wing Bat Muridae Hydromys chrysogaster Water Rat Muridae Mus musculus ^ House Mouse Muridae Rattus fuscipes Bush Rat Muridae Rattus lutreolus Swamp Rat Otariidae Arctocephalus pusillus doriferus ~ Australian Fur-seal Otariidae Arctocephalus forsteri ~ New Zealand Fur Seal Peramelidae Isoodon obesulus Southern Brown Bandicoot Peramelidae Perameles nasuta Long-nosed Bandicoot Petauridae Petaurus australis Yellow Bellied Glider Petauridae Petaurus breviceps Sugar Glider Phalangeridae Trichosurus cunninghami Mountain Brushtail Possum Phalangeridae Trichosurus vulpecula Common Brushtail Possum Phascolarctidae Phascolarctos cinereus Koala Potoroidae Potorous sp. # ~ Long-nosed or Long-footed Potoroo Pseudocheiridae Petauroides volans Greater Glider Pseudocheiridae Pseudocheirus peregrinus -

Mosquitoes This Is the Year of the Mosquito
Promoting an appreciation and understanding of insects and their relatives in the animal kingdom through public education and the development of an invertebrate education facility. Marchie’s Nursery is a great place to teach kids about insects! The Buzz . It is hard to believe it has been one year since we In the months ahead we’ll continue to build the wrote our first newsletter. What a year it has been! momentum that will be needed to reach our vision – We visited numerous classes and camps, expanded educating the public about insects and their relatives, our menagerie of bug ambassadors and brought them increasing community awareness and support, Marchie’s Nursery is a great place to teach kids about insects! to many community events, kept you up to date on searching for a location, expanding our circle of local insect sightings via Facebook, applied for and friends and supporters, and strengthening our received our 501(c)(3) non-profit status, grew our operations for the long-term. membership, began looking for a home for our We would like to thank everyone who has facility, and worked with a consultant to ready our supported our work by becoming a member, donating organization for the work ahead. Amongst all these time, following us on Facebook, or sharing their steps forward, this year also saw the loss of our friend enthusiasm for this vision. We wouldn’t be here and founding board member, Byron Weber. I know without you. he would be proud of our progress. ~ Jen Marangelo Insect In‐sight – Mosquitoes This is the year of the mosquito. -

Phylogeny and Evolution of Lepidoptera
EN62CH15-Mitter ARI 5 November 2016 12:1 I Review in Advance first posted online V E W E on November 16, 2016. (Changes may R S still occur before final publication online and in print.) I E N C N A D V A Phylogeny and Evolution of Lepidoptera Charles Mitter,1,∗ Donald R. Davis,2 and Michael P. Cummings3 1Department of Entomology, University of Maryland, College Park, Maryland 20742; email: [email protected] 2Department of Entomology, National Museum of Natural History, Smithsonian Institution, Washington, DC 20560 3Laboratory of Molecular Evolution, Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland 20742 Annu. Rev. Entomol. 2017. 62:265–83 Keywords Annu. Rev. Entomol. 2017.62. Downloaded from www.annualreviews.org The Annual Review of Entomology is online at Hexapoda, insect, systematics, classification, butterfly, moth, molecular ento.annualreviews.org systematics This article’s doi: Access provided by University of Maryland - College Park on 11/20/16. For personal use only. 10.1146/annurev-ento-031616-035125 Abstract Copyright c 2017 by Annual Reviews. Until recently, deep-level phylogeny in Lepidoptera, the largest single ra- All rights reserved diation of plant-feeding insects, was very poorly understood. Over the past ∗ Corresponding author two decades, building on a preceding era of morphological cladistic stud- ies, molecular data have yielded robust initial estimates of relationships both within and among the ∼43 superfamilies, with unsolved problems now yield- ing to much larger data sets from high-throughput sequencing. Here we summarize progress on lepidopteran phylogeny since 1975, emphasizing the superfamily level, and discuss some resulting advances in our understanding of lepidopteran evolution. -

MOTHS and BUTTERFLIES LEPIDOPTERA DISTRIBUTION DATA SOURCES (LEPIDOPTERA) * Detailed Distributional Information Has Been J.D
MOTHS AND BUTTERFLIES LEPIDOPTERA DISTRIBUTION DATA SOURCES (LEPIDOPTERA) * Detailed distributional information has been J.D. Lafontaine published for only a few groups of Lepidoptera in western Biological Resources Program, Agriculture and Agri-food Canada. Scott (1986) gives good distribution maps for Canada butterflies in North America but these are generalized shade Central Experimental Farm Ottawa, Ontario K1A 0C6 maps that give no detail within the Montane Cordillera Ecozone. A series of memoirs on the Inchworms (family and Geometridae) of Canada by McGuffin (1967, 1972, 1977, 1981, 1987) and Bolte (1990) cover about 3/4 of the Canadian J.T. Troubridge fauna and include dot maps for most species. A long term project on the “Forest Lepidoptera of Canada” resulted in a Pacific Agri-Food Research Centre (Agassiz) four volume series on Lepidoptera that feed on trees in Agriculture and Agri-Food Canada Canada and these also give dot maps for most species Box 1000, Agassiz, B.C. V0M 1A0 (McGugan, 1958; Prentice, 1962, 1963, 1965). Dot maps for three groups of Cutworm Moths (Family Noctuidae): the subfamily Plusiinae (Lafontaine and Poole, 1991), the subfamilies Cuculliinae and Psaphidinae (Poole, 1995), and ABSTRACT the tribe Noctuini (subfamily Noctuinae) (Lafontaine, 1998) have also been published. Most fascicles in The Moths of The Montane Cordillera Ecozone of British Columbia America North of Mexico series (e.g. Ferguson, 1971-72, and southwestern Alberta supports a diverse fauna with over 1978; Franclemont, 1973; Hodges, 1971, 1986; Lafontaine, 2,000 species of butterflies and moths (Order Lepidoptera) 1987; Munroe, 1972-74, 1976; Neunzig, 1986, 1990, 1997) recorded to date. -

Aravalli Range of Rajasthan and Special Thanks to Sh
Occasional Paper No. 353 Studies on Odonata and Lepidoptera fauna of foothills of Aravalli Range, Rajasthan Gaurav Sharma ZOOLOGICAL SURVEY OF INDIA OCCASIONAL PAPER NO. 353 RECORDS OF THE ZOOLOGICAL SURVEY OF INDIA Studies on Odonata and Lepidoptera fauna of foothills of Aravalli Range, Rajasthan GAURAV SHARMA Zoological Survey of India, Desert Regional Centre, Jodhpur-342 005, Rajasthan Present Address : Zoological Survey of India, M-Block, New Alipore, Kolkata - 700 053 Edited by the Director, Zoological Survey of India, Kolkata Zoological Survey of India Kolkata CITATION Gaurav Sharma. 2014. Studies on Odonata and Lepidoptera fauna of foothills of Aravalli Range, Rajasthan. Rec. zool. Surv. India, Occ. Paper No., 353 : 1-104. (Published by the Director, Zool. Surv. India, Kolkata) Published : April, 2014 ISBN 978-81-8171-360-5 © Govt. of India, 2014 ALL RIGHTS RESERVED . No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise without the prior permission of the publisher. This book is sold subject to the condition that it shall not, by way of trade, be lent, resold hired out or otherwise disposed of without the publisher’s consent, in any form of binding or cover other than that in which, it is published. The correct price of this publication is the price printed on this page. Any revised price indicated by a rubber stamp or by a sticker or by any other means is incorrect and should be unacceptable. PRICE Indian Rs. 800.00 Foreign : $ 40; £ 30 Published at the Publication Division by the Director Zoological Survey of India, M-Block, New Alipore, Kolkata - 700053 and printed at Calcutta Repro Graphics, Kolkata - 700 006. -

Pupae of Japanesecallidulidae(Lepidoptera)
The LepidopterologicalSocietyLepidopterological Society of Japan ue t ue Lapickvetera Science 62 (2): 98- 1Ol, June 201 1 Pupae of Japanese Callidulidae (Lepidoptera) Masanao NAKAMURA Ubumi 4971-1, Ytito-cho, Nishi-ku, Hamamatu, 431-O102 Japan Abstruct [[he pupae of two callidulid gellera, Callidula Hlibner and Ptervdecta Butler present in Japan are described. From the pupal characteristics, the Callidulidae as designated by Minet (1989) contains two (or more) conspicuously diEferent families, one (incEuding at least the Pterothysaninae) belongs to the Macrolepidoptera and is related to the Bombycoidea, the other (the Callidulinae) is a sister group ef the Hyblaeidae in the Microlepidoptera, True Callidu] Ldae should be restricted to Minet's Callidulinae, Key wordsCallidulidae, Callidula, Pterodecld, pupa, description, classification, phylogeny, Japan. The Callidulidae is a small but curious farnily. Minet separate fatnilies but also they are far apart in phylogenetic (1989) recognized three subfamilies, the Oriental relationships, the fOrmer is a member o'f the so-called Callidulinae, the Madagascan Griveaudiinae and the Microlepidoptera and the latter belongs within the higher Pterothysaninae.Arnongthem,onlytwogeneraPterodectaLepidoptera close to the Bombycoidea. and Callidula in the Callidulinae occur in Japan, Based on the pupal morphology, Nakarnura (1981) Knowledge of the pupae in this family is insufficient and considered R falderi Bren]er should be included that in pupue were recorded only three geilera so fai: Oriental the Pyraloidea -

Reward and Non-Reward Learning of Flower Colours in the Butterfly Byasa Alcinous (Lepidoptera: Papilionidae)
Naturwissenschaften (2012) 99:705–713 DOI 10.1007/s00114-012-0952-y ORIGINAL PAPER Reward and non-reward learning of flower colours in the butterfly Byasa alcinous (Lepidoptera: Papilionidae) Ikuo Kandori & Takafumi Yamaki Received: 17 January 2012 /Revised: 13 July 2012 /Accepted: 14 July 2012 /Published online: 1 August 2012 # Springer-Verlag 2012 Abstract Learning plays an important role in food acquisition social interaction and sexual behaviour (Dukas 2008). Learning for a wide range of insects. To increase their foraging efficien- is a fundamental mechanism for adjusting behaviour to envi- cy, flower-visiting insects may learn to associate floral cues ronmental change (Stephens 1993; Dunlap and Stephens 2009). with the presence (so-called reward learning) or the absence Although some aspects of learning are costly (e.g. Mery and (so-called non-reward learning) of a reward. Reward learning Kawecki 2003, 2004;Burgeretal.2008), learning generally whilst foraging for flowers has been demonstrated in many increasesfitness(DukasandBernays2000; Dukas and Duan insect taxa, whilst non-reward learning in flower-visiting 2000; Raine and Chittka 2008). Many studies have shown that insects has been demonstrated only in honeybees, bumblebees insects can develop a positive association between visual and/or and hawkmoths. This study examined both reward and non- olfactory cues and resources such as nectar or oviposition sites reward learning abilities in the butterfly Byasa alcinous whilst (e.g. Shafir 1996;Cnaanietal.2006;Dukas1999;Dukasand foraging among artificial flowers of different colours. This Bernays 2000; Weiss and Papaj 2003). This is often called butterfly showed both types of learning, although butterflies ‘positive (associative) learning’ (e.g. -
Moths Postfire to Feb16 2021 by Family
Species present after fire - by family Scientific Name Common Name Taxon Family Name Anthela acuta Common Anthelid Moth Anthelidae Anthela ferruginosa Anthelidae Anthela ocellata Eyespot Anthelid Moth Anthelidae Labdia chryselectra Cosmopterigidae Limnaecia sp. Cosmopterigidae Limnaecia camptosema Cosmopterigidae Macrobathra alternatella Cosmopterigidae Macrobathra astrota Cosmopterigidae Macrobathra leucopeda Cosmopterigidae Ptilomacra senex Cossidae Achyra affinitalis Cotton Web Spinner Crambidae Culladia cuneiferellus Crambidae Eudonia aphrodes Crambidae Hednota sp. Crambidae Hednota bivittella Crambidae Hednota pleniferellus Crambidae Heliothela ophideresana Crambidae Hellula hydralis cabbage centre grub Crambidae Hygraula nitens Pond moth Crambidae Metasia capnochroa Crambidae Metasia dicealis Crambidae Metasia liophaea Crambidae Musotima nitidalis Golden-brown Fern Moth Crambidae Musotima ochropteralis Australian maidenhair fern moth Crambidae Nacoleia oncophragma Crambidae Nacoleia rhoeoalis Crambidae Scoparia chiasta Crambidae Scoparia emmetropis Crambidae Scoparia exhibitalis Crambidae Scopariinae Moss-eating Crambid Snout Moths Crambidae Tipanaea patulella White Rush Moth Crambidae Agriophara confertella Depressariidae Eupselia beatella Depressariidae Eupselia carpocapsella Common Eupselia Moth Depressariidae Eutorna tricasis Depressariidae Peritropha oligodrachma Depressariidae Phylomictis sp. Depressariidae Amata nigriceps Erebidae Cyme structa Erebidae Dasypodia cymatodes Northern wattle moth Erebidae Dasypodia selenophora Southern -

Recent Literature on Lepidoptera
1956 The Lepidopterists' News 121 RECENT LITERATURE ON LEPIDOPTERA (Under the supervision of PBTER F. BELLINGER) Under this heading are included abstracts of papers and books of interest to lepidop terists. The world's literature is searched systematically, and it is intended that every work on Lepidoptera published after 1946 will be noticed here; omissions of papers more than 3 or 4 years old should be called to Dr. BELLINGER'S attention. New genera and higher categories are shown in CAPITALS, with types in parentheses; new species and subspecies are noted, with type localities if given in print, Larval food plants are usually listed. Critical comments by abstractors may be made. Papers of only local interest and papers from The Lepidopterists' News are listed without abstract. Readers, particularly outside of North America, interested in assisting with this very large task, are invited to write Dr. BELLINGER (Osborn Zoological Lab., Yale Universiry, New Haven 11, Conn., U. S. A.) Abstractors' initials are as follows: [P.B.] - P. F. BELLIN GER; (I.e.] - I. F. B. COMMON; [W.c.] - W. C. COOK; [A.D.] -A. DIAKONOFF; [W.H.]- W. HACKMAN; [J .M.]-]. MOUCHA; [E.M.] - E. G. MUNROE; [N.O.] N. S. OBRAZTSOV; [C.R.]- C. 1. REMINGTON; (J.T.]- J. W. TILDEN; [P.V.] P. E. 1. VIETTE. B. SYSTEMATICS AND NOMENCLATURE Bauer, David 1., "A new race of Papilio indra from the Grand Canyon region." Lepid. News, vo!.9: pp.49-54, 1 pI. 10 Aug. 1955. Describes as new p, i, kaibabemis (Bright Angel Point, Ariz.) Bell, Ernest Layton, & Cyril Franklin dos Passos, "The lectotype of Megathymus aryxna Dyar (Lepidoptera, Megathymidre)." Amer. -

Mountain Pine Beetle Mania
Mountain Pine Beetle mania A Junior High Science Resource Mountain Pine Beetle mania Resource Authors: John Cunnian, Inside Education Erin Gluck, Inside Education Steve McIsaac, Inside Education Resource Design: Kristin Manchakowski Technical Review: Mountain Pine Beetle Strategic Direction Council (See page 26 for contact information.) Inside Education www.insideeducation.ca [email protected] P: 780-421-1497 Th is teaching resource is supported by: Parks Canada Natural Resources Canada - Canadian Forest Service Alberta Sustainable Resource Development Alberta Community Development Hinton Wood Products - A Division of West Fraser Mills Ltd. Spray Lake Sawmills With additional support from: Alberta Newsprint Company Canfor Sundance Forest Industries Ltd. Weyerhaeuser Mountain Pine Beetle Curriculum Connections: Key Messages: Th is resource was designed to bring the biology and • Th e mountain pine beetle is a naturally occurring issues surrounding the mountain pine beetle into the insect found in pine forests in the southern Rocky spotlight for junior high science students in Alberta. Mountains and in areas west of the Continental Th e major curriculum connections are as follows: Divide; however, it has not historically occurred in the northeastern slopes of the Rocky Mountains. An Alberta Grade 7 Science Unit A: abundance of mature pine forests as a result of many Interactions & Ecosystems years of wildfi re suppression and milder winters have combined to enable the expansion of mountain pine Alberta Grade 7 Science Unit B: beetle into large tracts of pine forest — prime beetle Plants for Food & Fibre habitat. Alberta Grade 8 Science Unit B: • Th ere are a variety of diff erent groups concerned Cells & Systems about managing the mountain pine beetle. -

A LEPKESZET TORTENETE MAGYARORSZAGON Melyet Ut61)B, Pariz-Papai Szotara Szerint, «Selyemszar6 Fereg»
r * R .C.P. EDINBURGH LIBRARY R27821 Y0236 ABAFT AIGNER LAJOS. BUDAPEST. KIADJA A KIR. MAGYAR TERMKSZETTUDOMANYI TARSULAT 189S. f Az I S'.)5-ik tivi iiovemher :20-aii tartott valasztmanyi iile- sen Abafi Aigner Lajos tagtaisiink «A lepkeszet tortenete Magyarorszagon I) czimii dolgozatat felajaiilotta Tarsiilatuiik- nak kiadiiM vegett. A valasztinaiiy dr. Entz Geza es dr. Horvath (teza valasztmanyi tag urakat kerte fdl velemenyes jelentes ttdelere. A hirald maknak kedvezo jelentese alapjau a valasztmaiiy iSdd oktoher 21-iki iilesen a raiinkat kiadasra elfogadta, b a titkarsagot a dolgozatnak sajto ala rendezeBevel megbi'zta. E rnmika, kiadaBanak koltsegeit az orszagos segelybdl fedeztiik. JlndapeBt, iSdcS iiiajus 23-ikan. huzlavszky Jozsef a Kir. Magyar Termeszettudomauyi Tarsulat e. titkara. Digitized by the Internet Archive in 2016 https://archive.org/details/b21926256 ELOSZO. Aimi mondas, bogy «a tdrtenelem a tel tudasi), iiemcsak az egesz emberiseg vagy egyes nemzetek tdrtenelmere, baneina tudomanyok vagy azok barmel}'' aganak tdrtenetere vonatkozd- lag is ervenyes. A szakferbbra eji oly fontos, mint erdekes, liogy megtiidja, bogy a tudomanynak az az aga, melynek mii- kddeset szentelte, mikeiit fejlodott ; bogy azoii fertiak, kikiiek nevet az allatcsaladok, nemek s eg^^es fajok melle jegyezve latja, mennyil)en es mivel jarultak e tudoinanyag fejlesztesebez. Ennelfogva, midon a magyar lepkeszet tdrteneteiiek meg- irasara vallalkoztam, elsd sorban tiszta kepet kellett adnom annak, bogy a lepidopterologia altalaban mind fejlddesi sta- dinmokon ment keresztiil. Ez azonban nebez feladatiiak bizo- nyiilt, mive] a kiilfdldi irodalmak egyikeben sem talaljnk meg a lepidopterologia tdrtenetet. Az entomologia egesz kdrere nezve - egy ])ar regihh es csekelyebb erteku adatot bgyelmen kiviil bagyva egyediil Eiselt Np:p.